Numerische lineare Algebra
Die numerische lineare Algebra ist ein zentrales Teilgebiet der numerischen Mathematik. Sie beschäftigt sich mit der Entwicklung und der Analyse von Rechenverfahren (Algorithmen) für Problemstellungen der linearen Algebra, insbesondere der Lösung von linearen Gleichungssystemen und Eigenwertproblemen. Solche Probleme spielen in allen Natur- und Ingenieurwissenschaften, aber auch in der Ökonometrie und in der Statistik eine große Rolle.
Die Algorithmen der numerischen linearen Algebra lassen sich grob in zwei Gruppen einteilen: in die direkten Verfahren, die theoretisch nach endlich vielen Rechenschritten die exakte Lösung eines Problems liefern, und in die iterativen Verfahren, bei denen die exakte Lösung schrittweise immer genauer angenähert wird. Da aber auch die direkten Verfahren wegen der beim Rechnen mit endlicher Genauigkeit entstehenden Rundungsfehler nur Näherungen für die exakte Lösung liefern, ist diese Unterscheidung nur für die Entwicklung und Untersuchung der Verfahren selbst von Bedeutung; für den praktischen Einsatz spielt sie keine große Rolle. Historisch gehen die ersten systematischen Verfahren aus beiden Gruppen – das direkte gaußsche Eliminationsverfahren und das iterative Gauß-Seidel-Verfahren – auf Carl Friedrich Gauß zurück. Beispiele für bedeutende Verfahren des 20. Jahrhunderts, die zahlreiche Verbesserungen und Weiterentwicklungen zur Folge hatten, sind das Zerlegungsverfahren von André-Louis Cholesky, das QR-Verfahren für Eigenwertprobleme von John G. F. Francis und Wera Nikolajewna Kublanowskaja sowie das CG-Verfahren von Eduard Stiefel und Magnus Hestenes als erster Vertreter der wichtigen Krylow-Unterraum-Verfahren.
Einführung in die Problemstellungen
Ein – auch historisch gesehen – zentraler Anfangspunkt der
elementaren linearen Algebra sind lineare Gleichungssysteme. Wir betrachten
 Gleichungen der Gestalt
Gleichungen der Gestalt

für
Unbekannte  .
Die Koeffizienten
.
Die Koeffizienten  und
und  sind gegebene Zahlen; die gesuchten Werte für
sollen so bestimmt werden, dass alle Gleichungen erfüllt werden. Die
Koeffizienten lassen sich zu einer Matrix
sind gegebene Zahlen; die gesuchten Werte für
sollen so bestimmt werden, dass alle Gleichungen erfüllt werden. Die
Koeffizienten lassen sich zu einer Matrix
 zusammenfassen; die Zahlen
und die Unbekannten
zusammenfassen; die Zahlen
und die Unbekannten  bilden Spaltenvektoren
bilden Spaltenvektoren
 und
und  .
Auf diese Weise ergibt sich die Matrix-Vektor-Darstellung
.
Auf diese Weise ergibt sich die Matrix-Vektor-Darstellung
eines linearen Gleichungssystems: Gesucht ist ein Vektor  ,
der bei der Matrix-Vektor-Multiplikation
mit der gegebenen Matrix
,
der bei der Matrix-Vektor-Multiplikation
mit der gegebenen Matrix  den gegebenen Vektor
den gegebenen Vektor  ergibt. Als Teilgebiet der Numerik betrachtet auch die numerische lineare
Algebra nur sogenannte korrekt
gestellte Probleme, also insbesondere nur solche Probleme, die eine Lösung
besitzen und bei denen die Lösung eindeutig bestimmt ist. Insbesondere wird im
Folgenden stets angenommen, dass die Matrix
regulär
ist, also eine Inverse
ergibt. Als Teilgebiet der Numerik betrachtet auch die numerische lineare
Algebra nur sogenannte korrekt
gestellte Probleme, also insbesondere nur solche Probleme, die eine Lösung
besitzen und bei denen die Lösung eindeutig bestimmt ist. Insbesondere wird im
Folgenden stets angenommen, dass die Matrix
regulär
ist, also eine Inverse
 besitzt. Dann gibt es für jede rechte Seite
eine eindeutig bestimmte Lösung
des linearen Gleichungssystems, die formal als
besitzt. Dann gibt es für jede rechte Seite
eine eindeutig bestimmte Lösung
des linearen Gleichungssystems, die formal als  angegeben werden kann.
angegeben werden kann.
Viele wichtige Anwendungen führen allerdings auf lineare Gleichungssysteme
mit mehr Gleichungen als Unbekannten. In der Matrix-Vektor-Darstellung  hat in diesem Fall die Matrix
mehr Zeilen als Spalten. Solche überbestimmten Systeme haben im
Allgemeinen keine Lösung. Man behilft sich deshalb damit, den Vektor
so zu wählen, dass die Differenz
hat in diesem Fall die Matrix
mehr Zeilen als Spalten. Solche überbestimmten Systeme haben im
Allgemeinen keine Lösung. Man behilft sich deshalb damit, den Vektor
so zu wählen, dass die Differenz  ,
das Residuum,
in einem noch festzulegenden Sinn „möglichst klein“ wird. Beim mit Abstand
wichtigsten Fall, dem sogenannten linearen Ausgleichsproblem,
wird dazu die Methode
der kleinsten Quadrate verwendet: Hierbei wird
so gewählt, dass die Quadratsumme
,
das Residuum,
in einem noch festzulegenden Sinn „möglichst klein“ wird. Beim mit Abstand
wichtigsten Fall, dem sogenannten linearen Ausgleichsproblem,
wird dazu die Methode
der kleinsten Quadrate verwendet: Hierbei wird
so gewählt, dass die Quadratsumme  minimal wird, wobei
minimal wird, wobei  die Komponenten des Differenzvektors
die Komponenten des Differenzvektors  bezeichnen. Mithilfe der euklidischen
Norm lässt sich das auch so schreiben: Man wähle
so, dass
bezeichnen. Mithilfe der euklidischen
Norm lässt sich das auch so schreiben: Man wähle
so, dass  minimal wird.
minimal wird.
Neben den linearen Gleichungen sind die Eigenwertprobleme ein weiteres
zentrales Thema der linearen Algebra. Gegeben ist hierbei eine Matrix
mit
Zeilen und
Spalten; gesucht sind Zahlen  und Vektoren
und Vektoren  ,
sodass die Gleichung
,
sodass die Gleichung

erfüllt ist. Man nennt dann
einen Eigenvektor von
zum Eigenwert .
Das Problem, alle Eigenwerte und Eigenvektoren einer Matrix zu bestimmen, ist
gleichbedeutend damit sie zu diagonalisieren.
Das bedeutet: Man finde eine reguläre Matrix  und eine Diagonalmatrix
und eine Diagonalmatrix
 mit
mit  .
Die Diagonaleinträge von
sind dann die Eigenwerte von
und die Spalten von
die zugehörigen Eigenvektoren.
.
Die Diagonaleinträge von
sind dann die Eigenwerte von
und die Spalten von
die zugehörigen Eigenvektoren.
Diese Probleme treten in allen Natur- und Ingenieurwissenschaften auf. Sie spielen aber auch in den Wirtschaftswissenschaften sowie in der Statistik – und damit in allen Gebieten, die sich statistischer Methoden bedienen – eine große Rolle. Lineare Gleichungssysteme beschreiben beispielsweise Modelle in der Statik, elektrische Netzwerke oder volkswirtschaftliche Verflechtungen. So scheinbar unterschiedliche Aufgabenstellungen wie die Stabilitätsuntersuchung dynamischer Systeme, Resonanzphänomene bei Schwingungen, die Bestimmung eines PageRanks oder die Hauptkomponentenanalyse in der Statistik führen alle auf Eigenwertprobleme. Lineare Gleichungen entstehen auch durch Linearisierung und Diskretisierung innerhalb anderer numerischer Verfahren. So lassen sich beispielsweise zahlreiche Modelle in Naturwissenschaft und Technik durch partielle Differentialgleichungen beschreiben. Ihre numerische Lösung durch Differenzen- oder Finite-Elemente-Verfahren führt auf Systeme mit sehr vielen Unbekannten.
In diesem Übersichtsartikel wird der Einfachheit halber angenommen, dass alle gegebenen Matrizen und Vektoren reell sind, das heißt, dass alle ihre Einträge reelle Zahlen sind. Meist lassen sich die angesprochenen Verfahren direkt auf komplexe Zahlen verallgemeinern; an die Stelle der orthogonalen Matrizen tritt dann beispielsweise ihr komplexes Pendant, die unitären Matrizen. Mitunter ist es auch vorteilhaft, ein gegebenes komplexes Problem – etwa durch Betrachtung von Real- und Imaginärteil – auf ein reelles zurückzuführen. Zusatzüberlegungen treten allerdings bei Eigenwertproblemen mit nichtsymmetrischen reellen Matrizen auf, denn diese können auch nichtreelle Eigenwerte und Eigenvektoren haben.
Geschichte
Die Anfänge: Gauß und Jacobi

Bereits seit der Antike sind Lösungen konkreter Problemstellungen überliefert, die aus heutiger Sicht als lineare Gleichungssysteme angesehen werden können. Die Neun Kapitel der Rechenkunst, in denen der Stand der chinesischen Mathematik des 1. Jahrhunderts n. Chr. zusammengefasst ist, enthielten dabei bereits tabellarische Rechenvorschriften, die Eliminationsverfahren in Matrixdarstellung entsprachen. Die systematische Untersuchung linearer Gleichungssysteme setzte gegen Ende des 17. Jahrhunderts mit ihrer Formulierung mithilfe allgemeiner Koeffizienten ein. Nach Vorarbeiten von Gottfried Wilhelm Leibniz und Colin Maclaurin veröffentlichte Gabriel Cramer 1750 eine explizite Lösungsformel für beliebig viele Unbekannte mithilfe von Determinanten. Mit dieser cramerschen Regel war das Problem theoretisch vollständig gelöst, auch in Hinblick auf Existenz und Eindeutigkeit von Lösungen. Für deren praktische Berechnung erwies sich die Formel jedoch als völlig ungeeignet, weil der Rechenaufwand dabei mit der Anzahl der Unbekannten astronomisch schnell anwächst (siehe auch Cramersche Regel#Rechenaufwand).
Die erste Verwendung und Beschreibung systematischer Rechenverfahren für lineare Gleichungen geht auf Carl Friedrich Gauß (1777–1855) zurück. Zu Beginn des 19. Jahrhunderts waren die Bestimmung der Bahndaten astronomischer Objekte und die Landesvermessung durch Triangulation die wichtigsten Anwendungsaufgaben der mathematischen Praxis. 1801 erregte Gauß großes Aufsehen, als es ihm gelang, die Bahn des neu entdeckten Kleinplaneten Ceres aus wenigen Beobachtungen so genau zu bestimmen, dass Ceres Ende des Jahres wiedergefunden werden konnte. Für das zugehörige überbestimmte Gleichungssystem verwendete er die von ihm entdeckte Methode der kleinsten Quadrate. Das von ihm zur Berechnung der Lösung eingesetzte Eliminationsverfahren beschrieb Gauß systematisch ab 1809 im Rahmen der Bahnbestimmung des Asteroiden Pallas, allerdings noch direkt angewendet auf Quadratsummen.

Auch das erste Iterationsverfahren zur Lösung linearer Gleichungssysteme – das Gauß-Seidel-Verfahren – stammt von Gauß. In einem Brief an Christian Ludwig Gerling berichtete er 1823 von einem neuen einfachen Verfahren, mit dem die Lösung Schritt für Schritt immer besser angenähert werden könne. Gauß, der inzwischen mit der Triangulation des Königreichs Hannover beschäftigt war, schreibt darin, er rechne fast jeden Abend noch einen Iterationsschritt; das sei eine angenehme Abwechslung zur einförmigen Aufnahme der Messdaten. Das Verfahren sei so wenig anfällig für Fehler, dass es sich sogar „halb im Schlaf“ ausführen lasse. 1845 veröffentlichte Carl Gustav Jacob Jacobi ein anderes, ähnliches Iterationsverfahren, das Jacobi-Verfahren. Als Philipp Ludwig von Seidel, ein Schüler Jacobis, 1874 ein System mit 72 Unbekannten lösen musste, entwickelte er eine modifizierte, verbesserte Version dieser Methode. Wie sich im Nachhinein herausstellte, ist dieses Verfahren äquivalent zum Iterationsverfahren von Gauß, von dem Seidel jedoch vermutlich nichts wusste. Jacobi veröffentlichte 1846 auch ein iteratives Verfahren zur Transformation von Matrizen, das sich zur Lösung des Eigenwertproblems für symmetrische Matrizen eignet und heute ebenfalls als Jacobi-Verfahren bezeichnet wird. Er selbst verwendete es jedoch nur als Vorbereitungsschritt, um die Diagonaleinträge der Matrix stärker dominant zu machen.
20. Jahrhundert
Im Jahr 1923 wurde ein von André-Louis Cholesky entwickeltes Verfahren veröffentlicht, das bestimmte lineare Gleichungssysteme löst, indem die Koeffizientenmatrix in ein Produkt zweier einfacherer Matrizen zerlegt wird, die Cholesky-Zerlegung. Auch das gaußsche Eliminationsverfahren stellte sich im Nachhinein als ein Spezialfall solcher Matrixzerlegungsverfahren heraus. Algorithmen aus dieser Verfahrensgruppe sind auch heute noch die Standardverfahren zur Lösung mäßig großer Systeme.
Ab Ende der 1920er Jahre kamen auch neue Ideen zur iterativen Lösung von Eigenwertproblemen auf, beginnend 1929 mit der Vorstellung der Potenzmethode durch Richard von Mises. Wie auch bei der Weiterentwicklung zur inversen Iteration durch Helmut Wielandt 1944, können mit diesen einfachen Vektoriterationsverfahren immer nur Eigenvektoren zu einem einzelnen Eigenwert berechnet werden. Eine vollständige Lösung des Eigenwertproblems für beliebige Matrizen blieb aufwändig. Der Durchbruch kam hier 1961–1962 mit der Entwicklung des QR-Verfahrens durch den britischen Informatiker John G. F. Francis und unabhängig davon durch die russische Mathematikerin Wera Nikolajewna Kublanowskaja. Während Kublanowskaja in ihrer Arbeit von Anfang an ein tiefes Verständnis der Konvergenzeigenschaften der Methode aufzeigte, arbeitete Francis vor allem an Implementierungsdetails, die das Verfahren schnell und stabil machten. Das QR-Verfahren ist bis heute das Standardverfahren zur Berechnung aller Eigenwerte und Eigenvektoren nicht allzu großer Matrizen.

Die Lösung linearer Gleichungssysteme mit sehr großen Matrizen, wie sie bei der Diskretisierung von partiellen Differentialgleichungen auftreten, blieb weiterhin schwierig. Diese Matrizen haben nur relativ wenige Einträge, die ungleich null sind, und es ist von entscheidender Bedeutung, dass ein numerisches Verfahren diese Eigenschaft ausnutzt. Ein neuer Ansatz dazu, der sich als Startpunkt zahlreicher Weiterentwicklungen herausstellen sollte, war das 1952 von Eduard Stiefel und Magnus Hestenes entwickelte CG-Verfahren. Dabei wird das lineare Gleichungssystem in dem Spezialfall, dass die Matrix symmetrisch und zusätzlich positiv definit ist, durch ein äquivalentes Optimierungsproblem ersetzt. Als noch fruchtbarer erwies sich ein anderer Zugang zum CG-Verfahren, der gleichzeitig von Cornelius Lanczos entdeckt wurde: Die durch das CG-Verfahren berechneten Näherungen befinden sich in einer aufsteigenden Kette von Unterräumen, den Krylow-Räumen.
Trotz der Entdeckung dieser Zusammenhänge dauerte es relativ lange, bis konkrete Verallgemeinerungen des CG-Verfahrens entwickelt wurden. Das 1974 von Roger Fletcher veröffentlichte BiCG-Verfahren ist zwar theoretisch für beliebige Matrizen anwendbar, erwies sich jedoch in der Praxis in vielen Fällen als instabil. Das 1975 erschienene MINRES-Verfahren ist ein Krylow-Unterraum-Verfahren, für das die Matrix zwar symmetrisch sein muss, aber nicht unbedingt positiv definit wie beim CG-Verfahren. In der Folgezeit wurden zahlreiche Weiterentwicklungen untersucht, insbesondere Stabilisierungsversuche für das BiCG-Verfahren. Ein Beispiel für ein weit verbreitetes Krylow-Unterraum-Verfahren für beliebige lineare Gleichungssysteme ist eine Verallgemeinerung des MINRES-Verfahrens, das 1986 von Yousef Saad und Martin H. Schultz vorgestellte GMRES-Verfahren. Weitere Verfahren verwenden Synthesen aus Ideen der BiCG-Gruppe und GMRES, so das QMR-Verfahren (Roland W. Freund und Noel M. Nachtigal, 1991) sowie das TFQMR-Verfahren (Freund, 1993). Von Anfang an wurden Krylow-Unterraum-Verfahren auch zur Berechnung von Eigenwerten verwendet, Ausgangspunkte waren hier ein Verfahren von Lanczos 1950 und das Arnoldi-Verfahren von Walter Edwin Arnoldi 1951.
Grundprinzipien
“The field of numerical linear algebra is more beautiful, and more
fundamental, than its rather dull name may suggest. More beautiful, because it
is full of powerful ideas that are quite unlike those normally emphasized in a
linear algebra course in a mathematics department. […] More fundamental,
because, thanks to a trick of history, ‘numerical’ linear algebra is really
applied linear algebra.”
„Das Fachgebiet der numerischen linearen Algebra ist schöner und
grundlegender, als es sein ziemlich langweiliger Name vermuten lässt. Schöner,
weil es voll mächtiger Ideen ist, die ganz anders sind als diejenigen, die
normalerweise in einer Vorlesung über lineare Algebra an einem mathematischen
Institut als bedeutend herausgestellt werden. […] Grundlegender, weil
‚numerische‘ lineare Algebra dank eines Tricks der Geschichte in Wirklichkeit
angewandte lineare Algebra ist.“
Ausnutzung von Strukturen

Modelle und Fragestellungen in Wissenschaft und Technik können auf Probleme der linearen Algebra mit Millionen von Gleichungen führen. Die Einträge einer Matrix mit einer Million Zeilen und Spalten benötigen im double-precision-Format 8 Terabyte Speicherplatz. Das zeigt, dass bereits die Bereitstellung der Daten eines Problems, geschweige denn seine Lösung, eine Herausforderung darstellen, wenn nicht auch seine spezielle Struktur berücksichtigt wird. Glücklicherweise führen viele wichtige Anwendungen – wie beispielsweise die Diskretisierung partieller Differentialgleichungen mit der Finite-Elemente-Methode – zwar auf sehr viele Gleichungen, in jeder einzelnen Gleichung kommen jedoch nur relativ wenige Unbekannte vor. Für die zugehörige Matrix bedeutet das, dass es in jeder Zeile nur wenige Einträge ungleich null gibt, die Matrix ist, wie man sagt, dünnbesetzt. Es gibt zahlreiche Methoden, um solche Matrizen effizient abzuspeichern und ihre Struktur auszunutzen. Verfahren, in denen Matrizen nur in Matrix-Vektor-Produkten vorkommen, sind für dünnbesetzte Probleme besonders gut geeignet, da dabei alle Multiplikationen und Additionen mit null nicht explizit ausgeführt werden müssen. Algorithmen, bei denen die Matrix selbst umgeformt wird, sind hingegen meist nur schwierig zu implementieren, da dann die Dünnbesetztheit im Allgemeinen verloren geht.
Allgemein hat die Besetzungsstruktur, also die Anzahl und die Position der
Matrixeinträge ungleich null, einen sehr großen Einfluss auf die theoretischen
und numerischen Eigenschaften eines Problems. Das wird am Extremfall von Diagonalmatrizen, also
Matrizen, die nur auf der Hauptdiagonale Einträge ungleich null haben, besonders
deutlich. Ein lineares Gleichungssystem mit einer Diagonalmatrix kann einfach
gelöst werden, indem die Einträge auf der rechten Seite durch die
Diagonalelemente dividiert werden, also mittels
Divisionen. Auch lineare Ausgleichsprobleme und Eigenwertprobleme sind für
Diagonalmatrizen trivial. Die Eigenwerte einer Diagonalmatrix sind ihre
Diagonalelemente und die zugehörigen Eigenvektoren die Standardbasisvektoren
 .
.
Ein weiterer wichtiger Spezialfall sind die Dreiecksmatrizen, bei denen alle Einträge oberhalb oder unterhalb der Hauptdiagonale null sind. Gleichungssysteme mit solchen Matrizen können durch Vorwärts- bzw. Rückwärtseinsetzen einfach von oben nach unten bzw. von unten nach oben der Reihe nach aufgelöst werden. Die Eigenwerte von Dreiecksmatrizen sind wiederum trivialerweise die Einträge auf der Hauptdiagonale; zugehörige Eigenvektoren können ebenfalls durch Vorwärts- oder Rückwärtseinsetzen bestimmt werden. Ein weiterer häufiger Spezialfall dünnbesetzter Matrizen sind die Bandmatrizen: Hier sind nur die Hauptdiagonale und einige benachbarte Nebendiagonalen mit Einträgen ungleich null besetzt. Eine Abschwächung der oberen Dreiecksmatrizen sind die oberen Hessenbergmatrizen, bei den auch die Nebendiagonale unter der Hauptdiagonale besetzt ist. Eigenwertprobleme lassen sich mit relativ geringem Aufwand in äquivalente Probleme für Hessenberg- oder Tridiagonalmatrizen transformieren.
Aber nicht nur die Besetzungsstruktur, sondern auch andere Matrixeigenschaften spielen für die Entwicklung und Analyse numerischer Verfahren eine wichtige Rolle. Viele Anwendungen führen auf Probleme mit symmetrischen Matrizen. Insbesondere die Eigenwertprobleme sind deutlich einfacher zu handhaben, wenn die gegebene Matrix symmetrisch ist, aber auch bei linearen Gleichungssystemen reduziert sich in diesem Fall der Lösungsaufwand im Allgemeinen um etwa die Hälfte. Weitere Beispiele für Typen von Matrizen, für die spezialisierte Algorithmen existieren, sind die Vandermonde-Matrizen, die Toeplitz-Matrizen und die zirkulanten Matrizen.
Fehleranalyse: Vektor- und Matrixnormen
Als Maße für die „Größe“ eines Vektors  werden in der Mathematik unterschiedliche Vektornormen verwendet. Am
bekanntesten und verbreitetsten ist die euklidische
Norm
werden in der Mathematik unterschiedliche Vektornormen verwendet. Am
bekanntesten und verbreitetsten ist die euklidische
Norm
 ,
,
also die Wurzel aus der Summe der Quadrate aller Vektorkomponenten. Bei der
bekannten geometrischen Veranschaulichung von Vektoren als Pfeile im zwei- oder
dreidimensionalen Raum entspricht dies gerade der Pfeillänge. Je nach
untersuchter Fragestellung können jedoch auch andere Vektornormen wie etwa die
Maximumsnorm  oder die 1-Norm
oder die 1-Norm  geeigneter sein.
geeigneter sein.
Sind  Vektoren, wobei
Vektoren, wobei  als eine Näherung für
aufgefasst werden soll, so lässt sich mithilfe einer Vektornorm
als eine Näherung für
aufgefasst werden soll, so lässt sich mithilfe einer Vektornorm  die Genauigkeit dieser Näherung quantifizieren. Die Norm des Differenzvektors
die Genauigkeit dieser Näherung quantifizieren. Die Norm des Differenzvektors

wird als (normweiser) absoluter Fehler bezeichnet. Betrachtet man den
absoluten Fehler im Verhältnis zur Norm des „exakten“ Vektors
erhält man den (normweisen) relativen Fehler
 .
.
Da der relative Fehler nicht durch die Skalierung
von
und
beeinflusst wird, ist dieser das Standardmaß für den Unterschied der beiden
Vektoren und wird oft auch vereinfacht nur als „Fehler“ bezeichnet.
Auch die „Größe“ von Matrizen wird mit Normen
gemessen, den Matrixnormen.
Für die Wahl einer Matrixnorm  ist es wesentlich, dass sie zur verwendeten Vektornorm „passt“, insbesondere
soll die Ungleichung
ist es wesentlich, dass sie zur verwendeten Vektornorm „passt“, insbesondere
soll die Ungleichung  für alle
erfüllt sein. Definiert man
für eine gegebene Vektornorm als die kleinste Zahl
für alle
erfüllt sein. Definiert man
für eine gegebene Vektornorm als die kleinste Zahl  ,
sodass
,
sodass  für alle
gilt, dann erhält man die sogenannte natürliche
Matrixnorm. Für jede Vektornorm gibt es also eine davon induzierte
natürliche Matrixnorm: Für die euklidische Norm ist das die Spektralnorm
für alle
gilt, dann erhält man die sogenannte natürliche
Matrixnorm. Für jede Vektornorm gibt es also eine davon induzierte
natürliche Matrixnorm: Für die euklidische Norm ist das die Spektralnorm  ,
für die Maximumsnorm ist es die Zeilensummennorm
,
für die Maximumsnorm ist es die Zeilensummennorm
 und für die 1-Norm die Spaltensummennorm
und für die 1-Norm die Spaltensummennorm
 .
Analog zu Vektoren kann mithilfe einer Matrixnorm der relative Fehler
.
Analog zu Vektoren kann mithilfe einer Matrixnorm der relative Fehler

bei einer Näherung einer Matrix
durch eine Matrix  quantifiziert werden.
quantifiziert werden.
Kondition und Stabilität
Bei Problemen aus der Praxis sind gegebene Größen meist mit Fehlern behaftet,
den Datenfehlern. Zum Beispiel kann bei einem linearen Gleichungssystem
 die gegebene rechte Seite
aus einer Messung stammen und daher eine
Messabweichung
aufweisen. Aber auch bei theoretisch beliebig genau bekannten Größen lassen sich
Rundungsfehler bei ihrer
Darstellung im Computer als Gleitkommazahlen
nicht vermeiden. Es muss also davon ausgegangen werden, dass anstelle des
exakten Systems
in Wirklichkeit ein System
die gegebene rechte Seite
aus einer Messung stammen und daher eine
Messabweichung
aufweisen. Aber auch bei theoretisch beliebig genau bekannten Größen lassen sich
Rundungsfehler bei ihrer
Darstellung im Computer als Gleitkommazahlen
nicht vermeiden. Es muss also davon ausgegangen werden, dass anstelle des
exakten Systems
in Wirklichkeit ein System  mit einer gestörten rechten Seite
mit einer gestörten rechten Seite  und dementsprechend einer „falschen“ Lösung
vorliegt. Die grundlegende Frage ist nun, wie stark sich Störungen der gegebenen
Größen auf Störungen der gesuchten Größen auswirken. Wenn der relative Fehler
der Lösung nicht wesentlich größer ist als die relativen Fehler der
Eingangsgrößen, spricht man von einem gut
konditionierten, anderenfalls von einem schlecht konditionierten
Problem. Für das Beispiel linearer Gleichungssysteme lässt sich hierzu die
Abschätzung
und dementsprechend einer „falschen“ Lösung
vorliegt. Die grundlegende Frage ist nun, wie stark sich Störungen der gegebenen
Größen auf Störungen der gesuchten Größen auswirken. Wenn der relative Fehler
der Lösung nicht wesentlich größer ist als die relativen Fehler der
Eingangsgrößen, spricht man von einem gut
konditionierten, anderenfalls von einem schlecht konditionierten
Problem. Für das Beispiel linearer Gleichungssysteme lässt sich hierzu die
Abschätzung

beweisen. Das Problem ist also gut konditioniert, wenn  ,
das Produkt der Norm der Koeffizientenmatrix und der Norm ihrer Inversen, klein ist.
Diese wichtige Kenngröße heißt Konditionszahl
der Matrix
und wird mit
,
das Produkt der Norm der Koeffizientenmatrix und der Norm ihrer Inversen, klein ist.
Diese wichtige Kenngröße heißt Konditionszahl
der Matrix
und wird mit  bezeichnet. In realen Problemen wird meist nicht nur, wie hier dargestellt, die
rechte Seite
fehlerbehaftet sein, sondern auch die Matrix .
Dann gilt eine ähnliche, kompliziertere Abschätzung, in der aber ebenfalls
die wesentliche Kennzahl zur Bestimmung der Kondition des Problems bei kleinen
Datenfehlern ist.
Die Definition der Konditionszahl lässt sich auf nicht quadratische Matrizen
verallgemeinern und spielt dann auch eine wesentliche Rolle bei der Analyse
linearer Ausgleichsprobleme. Wie gut ein solches Problem konditioniert ist,
hängt allerdings nicht nur wie bei linearen Gleichungssystemen von der
Konditionszahl der Koeffizientenmatrix
ab, sondern auch von der rechten Seite ,
genauer vom Winkel
zwischen den Vektoren
bezeichnet. In realen Problemen wird meist nicht nur, wie hier dargestellt, die
rechte Seite
fehlerbehaftet sein, sondern auch die Matrix .
Dann gilt eine ähnliche, kompliziertere Abschätzung, in der aber ebenfalls
die wesentliche Kennzahl zur Bestimmung der Kondition des Problems bei kleinen
Datenfehlern ist.
Die Definition der Konditionszahl lässt sich auf nicht quadratische Matrizen
verallgemeinern und spielt dann auch eine wesentliche Rolle bei der Analyse
linearer Ausgleichsprobleme. Wie gut ein solches Problem konditioniert ist,
hängt allerdings nicht nur wie bei linearen Gleichungssystemen von der
Konditionszahl der Koeffizientenmatrix
ab, sondern auch von der rechten Seite ,
genauer vom Winkel
zwischen den Vektoren  und .
Nach dem Satz
von Bauer-Fike lässt sich auch die Kondition des Eigenwertproblems mit
Konditionszahlen beschreiben. Hier ist es jedoch nicht die Zahl ,
mit der sich Störungen der Eigenwerte abschätzen lassen, sondern
und .
Nach dem Satz
von Bauer-Fike lässt sich auch die Kondition des Eigenwertproblems mit
Konditionszahlen beschreiben. Hier ist es jedoch nicht die Zahl ,
mit der sich Störungen der Eigenwerte abschätzen lassen, sondern  ,
die Konditionszahl der Matrix ,
die
via
,
die Konditionszahl der Matrix ,
die
via  diagonalisiert.
diagonalisiert.
Während die Kondition eine Eigenschaft des zu lösenden Problems ist, ist Stabilität
eine Eigenschaft des dafür verwendeten Verfahrens. Ein numerischer Algorithmus
liefert – auch bei exakt gedachten Eingangsdaten – im Allgemeinen nicht die
exakte Lösung des Problems. Zum Beispiel muss ein iteratives Verfahren, das eine
wahre Lösung schrittweise immer genauer annähert, nach endlich vielen Schritten
mit der bis dahin erreichten Näherungslösung abbrechen. Aber auch bei direkten
Verfahren, die theoretisch in endlich vielen Rechenschritten die exakte Lösung
ergeben, kommt es bei der Umsetzung auf dem Computer bei jeder Rechenoperation
zu Rundungsfehlern. In der numerischen Mathematik werden zwei unterschiedliche
Stabilitätsbegriffe verwendet, die Vorwärtsstabilität und Rückwärtsstabilität.
Sei dazu allgemein  eine Eingabegröße eines Problems und
eine Eingabegröße eines Problems und  seine exakte Lösung, aufgefasst als Wert einer Funktion
seine exakte Lösung, aufgefasst als Wert einer Funktion  angewendet auf .
Auch wenn man die Eingabegröße als exakt vorgegeben betrachtet, wird die
Berechnung mit einem Algorithmus ein anderes, „falsches“ Ergebnis
angewendet auf .
Auch wenn man die Eingabegröße als exakt vorgegeben betrachtet, wird die
Berechnung mit einem Algorithmus ein anderes, „falsches“ Ergebnis  liefern, aufgefasst als Wert einer anderen, „falschen“ Funktion
liefern, aufgefasst als Wert einer anderen, „falschen“ Funktion  ebenfalls angewendet auf .
Ein Algorithmus heißt vorwärtsstabil, wenn sich
ebenfalls angewendet auf .
Ein Algorithmus heißt vorwärtsstabil, wenn sich  nicht wesentlich stärker von
nicht wesentlich stärker von  unterscheidet, als es aufgrund der Fehler in der Eingangsgröße
und der Kondition des Problems sowieso zu erwarten wäre.
Mit einer formalen Definition dieses Begriffs erhält man zwar ein naheliegendes
und relativ anschauliches Maß für die Stabilität, aber bei komplizierten
Algorithmen ist es oft schwierig, ihre Vorwärtsstabilität zu untersuchen. Daher
wird im Allgemeinen nach einer Idee von James
H. Wilkinson zunächst eine sogenannte Rückwärtsanalyse betrachtet: Dazu wird
ein
unterscheidet, als es aufgrund der Fehler in der Eingangsgröße
und der Kondition des Problems sowieso zu erwarten wäre.
Mit einer formalen Definition dieses Begriffs erhält man zwar ein naheliegendes
und relativ anschauliches Maß für die Stabilität, aber bei komplizierten
Algorithmen ist es oft schwierig, ihre Vorwärtsstabilität zu untersuchen. Daher
wird im Allgemeinen nach einer Idee von James
H. Wilkinson zunächst eine sogenannte Rückwärtsanalyse betrachtet: Dazu wird
ein  bestimmt mit
bestimmt mit  ,
das heißt: Der durch das Verfahren berechnete „falsche“ Wert wird aufgefasst als
„richtiger“ Wert, der aber mit einem anderen Wert der Eingabegröße berechnet
wurde.
Ein Algorithmus heißt rückwärtsstabil, wenn sich
nicht wesentlich stärker von
unterscheidet, als es aufgrund der Fehler in dieser Eingangsgröße sowieso zu
erwarten wäre. Es lässt sich beweisen, dass ein rückwärtsstabiler Algorithmus
auch vorwärtsstabil ist.
,
das heißt: Der durch das Verfahren berechnete „falsche“ Wert wird aufgefasst als
„richtiger“ Wert, der aber mit einem anderen Wert der Eingabegröße berechnet
wurde.
Ein Algorithmus heißt rückwärtsstabil, wenn sich
nicht wesentlich stärker von
unterscheidet, als es aufgrund der Fehler in dieser Eingangsgröße sowieso zu
erwarten wäre. Es lässt sich beweisen, dass ein rückwärtsstabiler Algorithmus
auch vorwärtsstabil ist.
Orthogonalität und orthogonale Matrizen
Wie die lineare Algebra zeigt, besteht ein enger Zusammenhang zwischen
Matrizen und Basen
des Vektorraums  .
Sind
linear
unabhängige Vektoren
.
Sind
linear
unabhängige Vektoren  im
gegeben, so sind diese eine Basis des Raums und jeder andere Vektor kann
eindeutig als Linearkombination
der Basisvektoren dargestellt werden. Ein Basiswechsel
entspricht dabei der Multiplikation gegebener Vektoren und Matrizen mit einer
Transformationsmatrix. Einen wichtigen Spezialfall bilden die Orthonormalbasen.
Hierbei sind die Basisvektoren paarweise orthogonal
zueinander („stehen senkrecht aufeinander“) und sind zudem alle auf euklidische Länge 1
normiert, so wie die Standardbasis
im
gegeben, so sind diese eine Basis des Raums und jeder andere Vektor kann
eindeutig als Linearkombination
der Basisvektoren dargestellt werden. Ein Basiswechsel
entspricht dabei der Multiplikation gegebener Vektoren und Matrizen mit einer
Transformationsmatrix. Einen wichtigen Spezialfall bilden die Orthonormalbasen.
Hierbei sind die Basisvektoren paarweise orthogonal
zueinander („stehen senkrecht aufeinander“) und sind zudem alle auf euklidische Länge 1
normiert, so wie die Standardbasis
 im dreidimensionalen Raum. Fasst man die Basisvektoren spaltenweise zu einer
Matrix
im dreidimensionalen Raum. Fasst man die Basisvektoren spaltenweise zu einer
Matrix

zusammen, so erhält man im Fall einer Orthonormalbasis eine sogenannte orthogonale Matrix.
Orthonormalbasen und orthogonale Matrizen besitzen zahlreiche bemerkenswerte
Eigenschaften, auf denen die wichtigsten Verfahren der modernen numerischen
linearen Algebra basieren.
Die Tatsache, dass bei einer orthogonalen Matrix  die Spalten eine Orthonormalbasis bilden, lässt sich in Matrixschreibweise durch
die Gleichung
die Spalten eine Orthonormalbasis bilden, lässt sich in Matrixschreibweise durch
die Gleichung  ausdrücken, wobei
ausdrücken, wobei  die transponierte
Matrix und
die transponierte
Matrix und  die Einheitsmatrix
bezeichnen. Das zeigt wiederum, dass eine orthogonale Matrix regulär ist und
ihre Inverse
gleich ihrer Transponierten ist:
die Einheitsmatrix
bezeichnen. Das zeigt wiederum, dass eine orthogonale Matrix regulär ist und
ihre Inverse
gleich ihrer Transponierten ist:  .
Die Lösung eines linearen Gleichungssystems
.
Die Lösung eines linearen Gleichungssystems  lässt sich daher sehr einfach bestimmen, es gilt
lässt sich daher sehr einfach bestimmen, es gilt  .
Eine andere grundlegende Eigenschaft ist es, dass eine Multiplikation eines
Vektors mit einer orthogonalen Matrix seine euklidische Norm unverändert lässt
.
Eine andere grundlegende Eigenschaft ist es, dass eine Multiplikation eines
Vektors mit einer orthogonalen Matrix seine euklidische Norm unverändert lässt
 .
.
Damit folgt für die Spektralnorm  und für die Konditionszahl ebenfalls
und für die Konditionszahl ebenfalls
 ,
,
denn
ist ebenfalls eine orthogonale Matrix. Multiplikationen mit orthogonalen
Matrizen bewirken also keine Vergrößerung des relativen Fehlers.
Orthogonale Matrizen spielen auch eine wichtige Rolle in der Theorie und der
numerischen Behandlung von Eigenwertproblemen. Nach der einfachsten Version des
Spektralsatzes lassen sich
symmetrische Matrizen orthogonal diagonalisieren. Damit ist gemeint: Zu einer
Matrix ,
für die  gilt, existiert eine orthogonale Matrix
und eine Diagonalmatrix
mit
gilt, existiert eine orthogonale Matrix
und eine Diagonalmatrix
mit
 .
.
Auf der Diagonale von
stehen die Eigenwerte von
und die Spalten von
bilden eine Orthonormalbasis aus Eigenvektoren. Insbesondere ist nach dem oben
erwähnten Satz von Bauer-Fike das Eigenwertproblem für symmetrische Matrizen
stets gut konditioniert.
Mit der sogenannten schurschen
Normalform existiert eine Verallgemeinerung dieser orthogonalen
Transformation für nichtsymmetrische Matrizen.
Es gibt zwei spezielle, leicht handhabbare Arten orthogonaler Matrizen, die in zahllosen konkreten Verfahren der numerischen linearen Algebra zum Einsatz kommen: die Householder-Matrizen und die Givens-Rotationen. Householder-Matrizen haben die Gestalt
mit einem Vektor  mit
mit  .
Geometrisch beschreiben sie Spiegelungen
des -dimensionalen
Raums
an der
.
Geometrisch beschreiben sie Spiegelungen
des -dimensionalen
Raums
an der  -dimensionalen
Hyperebene durch den
Nullpunkt, die orthogonal zu
-dimensionalen
Hyperebene durch den
Nullpunkt, die orthogonal zu  ist. Ihre wesentliche Eigenschaft ist die folgende: Zu einem gegebenen Vektor
ist. Ihre wesentliche Eigenschaft ist die folgende: Zu einem gegebenen Vektor
 lässt sich leicht ein Vektor
bestimmen, sodass die zugehörige Householder-Matrix
lässt sich leicht ein Vektor
bestimmen, sodass die zugehörige Householder-Matrix  den Vektor
auf ein Vielfaches von
den Vektor
auf ein Vielfaches von  transformiert:
transformiert:  mit
mit  .
Dieses
.
Dieses  transformiert also alle Einträge von
bis auf den ersten zu null. Wendet man auf diese Weise geeignete
Householder-Transformationen Spalte für Spalte nacheinander auf eine Matrix
an, so können alle Einträge von
unterhalb der Hauptdiagonale zu null transformiert werden.
transformiert also alle Einträge von
bis auf den ersten zu null. Wendet man auf diese Weise geeignete
Householder-Transformationen Spalte für Spalte nacheinander auf eine Matrix
an, so können alle Einträge von
unterhalb der Hauptdiagonale zu null transformiert werden.
Givens-Rotationen sind spezielle Drehungen
des ,
die eine zweidimensionale Ebene drehen und die anderen  Dimensionen fest lassen. Die Transformation eines Vektors
mit einer Givens-Rotation verändert daher nur zwei Einträge von .
Durch geeignete Wahl des Drehwinkels kann dabei einer der beiden Einträge auf
null gesetzt wird. Während Householder-Transformationen, angewendet auf
Matrizen, ganze Teilspalten transformieren, können Givens-Rotationen dazu
verwendet werden, gezielt einzelne Matrixeinträge zu ändern.
Dimensionen fest lassen. Die Transformation eines Vektors
mit einer Givens-Rotation verändert daher nur zwei Einträge von .
Durch geeignete Wahl des Drehwinkels kann dabei einer der beiden Einträge auf
null gesetzt wird. Während Householder-Transformationen, angewendet auf
Matrizen, ganze Teilspalten transformieren, können Givens-Rotationen dazu
verwendet werden, gezielt einzelne Matrixeinträge zu ändern.
Householder-Transformationen und Givens-Rotationen können also dazu benutzt
werden, eine gegebene Matrix
auf eine obere Dreiecksmatrix zu transformieren, oder anders ausgedrückt, eine
QR-Zerlegung  in eine orthogonale Matrix und eine obere Dreiecksmatrix zu berechnen. Die
QR-Zerlegung ist ein wichtiges und vielseitiges Werkzeug, das in zahlreichen
Verfahren aus allen Bereichen der numerischen linearen Algebra zum Einsatz
kommt.
in eine orthogonale Matrix und eine obere Dreiecksmatrix zu berechnen. Die
QR-Zerlegung ist ein wichtiges und vielseitiges Werkzeug, das in zahlreichen
Verfahren aus allen Bereichen der numerischen linearen Algebra zum Einsatz
kommt.
Ähnlichkeitstransformationen
In der linearen Algebra wird zur Untersuchung des Eigenwertproblems  einer Matrix
mit
Zeilen und
Spalten das charakteristische
Polynom
einer Matrix
mit
Zeilen und
Spalten das charakteristische
Polynom  verwendet, ein Polynom vom Grad .
Die Eigenwerte von
sind genau die Nullstellen
von
verwendet, ein Polynom vom Grad .
Die Eigenwerte von
sind genau die Nullstellen
von  .
Mit dem Fundamentalsatz
der Algebra ergibt sich daraus direkt, dass
genau
Eigenwerte besitzt, wenn sie mit ihrer Vielfachheit
gezählt werden. Allerdings können diese Eigenwerte, auch bei reellen Matrizen,
komplexe Zahlen sein. Ist jedoch
eine reelle symmetrische Matrix, dann sind ihre Eigenwerte alle reell.
.
Mit dem Fundamentalsatz
der Algebra ergibt sich daraus direkt, dass
genau
Eigenwerte besitzt, wenn sie mit ihrer Vielfachheit
gezählt werden. Allerdings können diese Eigenwerte, auch bei reellen Matrizen,
komplexe Zahlen sein. Ist jedoch
eine reelle symmetrische Matrix, dann sind ihre Eigenwerte alle reell.
Das charakteristische Polynom hat zwar eine große theoretische Bedeutung für
das Eigenwertproblem, zur numerischen Berechnung ist es jedoch nicht geeignet.
Das liegt vor allem daran, dass das Problem, aus gegebenen Koeffizienten die
Nullstellen des zugehörigen Polynoms zu berechnen, im Allgemeinen sehr schlecht
konditioniert ist: Kleine Störungen wie Rundungsfehler an Koeffizienten eines
Polynoms können zu einer starken Verschiebung seiner Nullstellen führen. Damit
würde ein gegebenenfalls gut konditioniertes Problem – die Berechnung der
Eigenwerte – durch ein zwar mathematisch äquivalentes, aber schlecht
konditioniertes Problem – die Berechnung der Nullstellen des charakteristischen
Polynoms – ersetzt.
Viele numerische Verfahren zur Berechnung von Eigenwerten und Eigenvektoren
beruhen daher auf einer anderen Grundidee, den Ähnlichkeitstransformationen:
Zwei quadratische Matrizen
und  werden ähnlich
genannt, wenn es eine reguläre Matrix
mit
werden ähnlich
genannt, wenn es eine reguläre Matrix
mit

gibt. Es kann gezeigt werden, dass zueinander ähnliche Matrizen die gleichen
Eigenwerte haben, bei einer Ähnlichkeitstransformation der Matrix
auf die Matrix
ändern sich also die gesuchten Eigenwerte nicht. Auch die zugehörigen
Eigenvektoren lassen sich leicht ineinander umrechnen: Ist
ein Eigenvektor von ,
dann ist  ein Eigenvektor von
zum gleichen Eigenwert. Das führt zu Grundideen, die in zahlreichen Algorithmen
zum Einsatz kommen. Die Matrix
wird durch Ähnlichkeitstransformation in eine Matrix überführt, für die das
Eigenwertproblem effizienter zu lösen ist, oder es wird eine Folge von
Ähnlichkeitstransformationen konstruiert, bei denen sich die Matrix einer
Diagonal- oder Dreiecksmatrix immer weiter annähert. Aus den oben genannten
Gründen werden dabei für die Transformationsmatrizen
meist orthogonale Matrizen verwendet.
ein Eigenvektor von
zum gleichen Eigenwert. Das führt zu Grundideen, die in zahlreichen Algorithmen
zum Einsatz kommen. Die Matrix
wird durch Ähnlichkeitstransformation in eine Matrix überführt, für die das
Eigenwertproblem effizienter zu lösen ist, oder es wird eine Folge von
Ähnlichkeitstransformationen konstruiert, bei denen sich die Matrix einer
Diagonal- oder Dreiecksmatrix immer weiter annähert. Aus den oben genannten
Gründen werden dabei für die Transformationsmatrizen
meist orthogonale Matrizen verwendet.
Verfahren und Verfahrensklassen
Gaußsches Eliminationsverfahren
Das klassische Eliminationsverfahren von Gauß zur Lösung linearer Gleichungssysteme ist ein Ausgangspunkt und Vergleichsmaßstab für weiterentwickelte Verfahren. Es wird aber auch immer noch als einfaches und zuverlässiges Verfahren – insbesondere in seiner Modifikation als LR-Zerlegung (siehe unten) – für nicht zu große, gut konditionierte Systeme in der Praxis verbreitet eingesetzt. Das Verfahren eliminiert systematisch Variablen aus den gegebenen Gleichungen, indem geeignete Vielfache einer Gleichung von einer anderen Gleichung subtrahiert werden, bis ein System in Stufenform entsteht, das der Reihe nach von unten nach oben aufgelöst werden kann.
Numerische Überlegungen kommen ins Spiel, wenn die Stabilität des Verfahrens
betrachtet wird. Soll mit dem  -ten
Diagonalelement
-ten
Diagonalelement  der Matrix
ein Element
der Matrix
ein Element  in derselben Spalte eliminiert werden, dann muss mit dem Quotienten
in derselben Spalte eliminiert werden, dann muss mit dem Quotienten

das  -fache
der -ten
Zeile von der
-fache
der -ten
Zeile von der  -Zeile
subtrahiert werden. Dazu muss zumindest
-Zeile
subtrahiert werden. Dazu muss zumindest  gelten, was sich durch geeignete Zeilenvertauschungen für eine reguläre Matrix
stets erreichen lässt. Aber mehr noch: Ist
gelten, was sich durch geeignete Zeilenvertauschungen für eine reguläre Matrix
stets erreichen lässt. Aber mehr noch: Ist  sehr klein im Vergleich zu
sehr klein im Vergleich zu  ,
dann ergäbe sich ein sehr großer Betrag von .
In den nachfolgenden Schritten bestünde dann die Gefahr von Stellenauslöschungen
durch Subtraktionen großer Zahlen und das Verfahren wäre instabil. Daher ist es
wichtig, durch Zeilenvertauschungen, sogenannte Pivotisierung, dafür zu
sorgen, dass die Beträge
,
dann ergäbe sich ein sehr großer Betrag von .
In den nachfolgenden Schritten bestünde dann die Gefahr von Stellenauslöschungen
durch Subtraktionen großer Zahlen und das Verfahren wäre instabil. Daher ist es
wichtig, durch Zeilenvertauschungen, sogenannte Pivotisierung, dafür zu
sorgen, dass die Beträge  möglichst klein bleiben.
möglichst klein bleiben.
Faktorisierungsverfahren
Die wichtigsten direkten Verfahren zur Lösung linearer Gleichungssysteme
lassen sich als Faktorisierungsverfahren darstellen. Deren Grundidee ist es, die
Koeffizientenmatrix
des Systems
in ein Produkt aus zwei oder mehr Matrizen zu zerlegen, allgemein etwa  .
Das lineare Gleichungssystem lautet damit
.
Das lineare Gleichungssystem lautet damit  und wird in zwei Schritten gelöst: Zuerst wird die Lösung
und wird in zwei Schritten gelöst: Zuerst wird die Lösung  des Systems
des Systems  berechnet und anschließend die Lösung
des Systems
berechnet und anschließend die Lösung
des Systems  .
Es gilt dann
.
Es gilt dann  ,
also ist
die Lösung des ursprünglichen Problems. Auf den ersten Blick scheint dabei nur
die Aufgabe, ein lineares Gleichungssystem zu lösen, durch die Aufgabe,
zwei lineare Gleichungssysteme zu lösen, ersetzt zu werden. Die Idee
dahinter ist es jedoch, die Faktoren
und
,
also ist
die Lösung des ursprünglichen Problems. Auf den ersten Blick scheint dabei nur
die Aufgabe, ein lineares Gleichungssystem zu lösen, durch die Aufgabe,
zwei lineare Gleichungssysteme zu lösen, ersetzt zu werden. Die Idee
dahinter ist es jedoch, die Faktoren
und  so zu wählen, dass die beiden Teilsysteme wesentlich einfacher zu lösen sind als
das Ausgangssystem. Ein offensichtlicher Vorteil der Verfahrensklasse ergibt
sich im Fall, dass mehrere lineare Gleichungssysteme mit derselben
Koeffizientenmatrix ,
aber unterschiedlichen rechten Seiten gelöst werden sollen: Die Faktorisierung
von ,
im Allgemeinen der aufwändigste Verfahrensschritt, muss dann nur einmal
berechnet werden.
so zu wählen, dass die beiden Teilsysteme wesentlich einfacher zu lösen sind als
das Ausgangssystem. Ein offensichtlicher Vorteil der Verfahrensklasse ergibt
sich im Fall, dass mehrere lineare Gleichungssysteme mit derselben
Koeffizientenmatrix ,
aber unterschiedlichen rechten Seiten gelöst werden sollen: Die Faktorisierung
von ,
im Allgemeinen der aufwändigste Verfahrensschritt, muss dann nur einmal
berechnet werden.
LR-Zerlegung
Das gaußsche Eliminationsverfahren kann als Faktorisierungsverfahren
aufgefasst werden. Trägt man die Koeffizienten
für  in eine Matrix ein, ergibt sich ohne Zeilenvertauschungen
in eine Matrix ein, ergibt sich ohne Zeilenvertauschungen  mit einer unteren Dreiecksmatrix
und einer oberen Dreiecksmatrix
mit einer unteren Dreiecksmatrix
und einer oberen Dreiecksmatrix  .
Zusätzlich ist
unipotent,
das heißt alle Einträge auf der Hauptdiagonale von
sind gleich 1. Wie gesehen müssen im Allgemeinen bei der Gauß-Elimination Zeilen
von
vertauscht werden. Das lässt sich formal mit Hilfe einer Permutationsmatrix
.
Zusätzlich ist
unipotent,
das heißt alle Einträge auf der Hauptdiagonale von
sind gleich 1. Wie gesehen müssen im Allgemeinen bei der Gauß-Elimination Zeilen
von
vertauscht werden. Das lässt sich formal mit Hilfe einer Permutationsmatrix
 darstellen, indem anstelle von
die zeilenpermutierte Matrix
darstellen, indem anstelle von
die zeilenpermutierte Matrix  faktorisiert wird:
faktorisiert wird:
 .
.
Nach dem Grundprinzip der Faktorisierungsverfahren werden zur Lösung von
also zunächst wie beschrieben die Dreiecksmatrizen
und
sowie gegebenenfalls die zugehörige Permutation bestimmt. In nächsten Schritt
wird  mit der zeilenpermutierten rechten Seite durch Vorwärtseinsetzen und schließlich
mit der zeilenpermutierten rechten Seite durch Vorwärtseinsetzen und schließlich
 durch Rückwärtseinsetzen gelöst.
durch Rückwärtseinsetzen gelöst.
Die LR-Zerlegung und damit das gaußsche Eliminationsverfahren ist mit geeigneter Pivotisierung „fast immer stabil“, das heißt in den meisten praktischen Anwendungsaufgaben tritt keine große Fehlerverstärkung auf. Es lassen sich jedoch pathologische Beispiele angeben, bei denen die Verfahrensfehler exponentiell mit der Anzahl der Unbekannten anwachsen.
Cholesky-Zerlegung
Die Cholesky-Zerlegung ist wie die LR-Zerlegung eine Faktorisierung der
Matrix
in zwei Dreiecksmatrizen für den in vielen Anwendungen auftretenden Fall, dass
symmetrisch und positiv
definit ist, also
erfüllt und nur positive Eigenwerte besitzt. Unter diesen Voraussetzungen gibt
es eine untere Dreiecksmatrix
mit
 .
.
Ein allgemeiner Ansatz für die Matrixeinträge von
führt auf ein explizites Verfahren, mit dem diese spaltenweise oder zeilenweise
nacheinander berechnet werden können, das Cholesky-Verfahren. Durch diese
Ausnutzung der Symmetrie von
reduziert sich der Rechenaufwand gegenüber der LR-Zerlegung auf etwa die
Hälfte.
Symmetrische und positiv definite Koeffizientenmatrizen treten klassisch bei
der Formulierung der sogenannten Normalgleichungen zur
Lösung linearer Ausgleichsprobleme auf. Man kann zeigen, dass das Problem,  zu minimieren, äquivalent damit ist, das lineare Gleichungssystem
zu minimieren, äquivalent damit ist, das lineare Gleichungssystem

zu lösen. Die Koeffizientenmatrix  dieser Normalgleichungen ist symmetrisch und, wenn die Spalten von
linear unabhängig sind, auch positiv definit. Es kann also mit dem
Cholesky-Verfahren gelöst werden.
Dieses Vorgehen empfiehlt sich jedoch nur für gut konditionierte Probleme mit
wenigen Unbekannten. Im Allgemeinen ist nämlich das System der Normalgleichungen
deutlich schlechter konditioniert als das ursprünglich gegebene lineare
Ausgleichsproblem. Es ist dann besser, nicht den Umweg über die
Normalgleichungen zu gehen, sondern direkt eine QR-Zerlegung von
zu verwenden.
dieser Normalgleichungen ist symmetrisch und, wenn die Spalten von
linear unabhängig sind, auch positiv definit. Es kann also mit dem
Cholesky-Verfahren gelöst werden.
Dieses Vorgehen empfiehlt sich jedoch nur für gut konditionierte Probleme mit
wenigen Unbekannten. Im Allgemeinen ist nämlich das System der Normalgleichungen
deutlich schlechter konditioniert als das ursprünglich gegebene lineare
Ausgleichsproblem. Es ist dann besser, nicht den Umweg über die
Normalgleichungen zu gehen, sondern direkt eine QR-Zerlegung von
zu verwenden.
QR-Zerlegung
Das lineare Gleichungssystem
kann nach der Berechnung einer QR-Zerlegung
direkt nach dem allgemeinen Prinzip der Faktorisierungsverfahren gelöst
werden; es ist nur noch
mit  durch Rückwärtseinsetzen zu bestimmen. Aufgrund der guten Kondition orthogonaler
Matrizen treten dabei die möglichen Instabilitäten der LR-Zerlegung nicht
ein.
Allerdings ist der Rechenaufwand im Allgemeinen etwa doppelt so groß, sodass
unter Umständen eine Abwägung der Verfahren getroffen werden muss.
durch Rückwärtseinsetzen zu bestimmen. Aufgrund der guten Kondition orthogonaler
Matrizen treten dabei die möglichen Instabilitäten der LR-Zerlegung nicht
ein.
Allerdings ist der Rechenaufwand im Allgemeinen etwa doppelt so groß, sodass
unter Umständen eine Abwägung der Verfahren getroffen werden muss.
Die QR-Zerlegung ist auch das gängige Verfahren zur Lösung nicht zu großer, gut konditionierter linearer Ausgleichsprobleme. Für das Problem
- Minimiere
gilt mit
und 
 .
.
Dabei wurde verwendet, dass
orthogonal ist, also die euklidische Norm erhält, und dass
gilt. Der letzte Ausdruck lässt sich einfach durch Rückwärtseinsetzen der ersten
Zeilen von  minimieren.
minimieren.
Fixpunktiteration mit Splitting-Verfahren
Eine völlig andere Idee, um
zu lösen, besteht darin, einen Startvektor  zu wählen und daraus schrittweise
zu wählen und daraus schrittweise  ,
,
 immer neue Näherungen an die gesuchte Lösung zu berechnen. Im Fall der Konvergenz der Folge
immer neue Näherungen an die gesuchte Lösung zu berechnen. Im Fall der Konvergenz der Folge
 gegen
wird dann diese Iteration
nach einer geeigneten Anzahl
gegen
wird dann diese Iteration
nach einer geeigneten Anzahl  von Schritten mit einer ausreichend genauen Näherung
von Schritten mit einer ausreichend genauen Näherung  für
abgebrochen. Die einfachsten und wichtigsten Verfahren dieser Art verwenden eine
Iteration der Gestalt
für
abgebrochen. Die einfachsten und wichtigsten Verfahren dieser Art verwenden eine
Iteration der Gestalt
mit einer geeigneten Matrix  und einem geeigneten Vektor
und einem geeigneten Vektor  .
Es lässt sich beweisen, dass solche Verfahren genau dann konvergieren, wenn
alle Eigenwerte von
einen Betrag echt kleiner als 1 haben. In diesem Fall konvergieren die
Iterierten
.
Es lässt sich beweisen, dass solche Verfahren genau dann konvergieren, wenn
alle Eigenwerte von
einen Betrag echt kleiner als 1 haben. In diesem Fall konvergieren die
Iterierten  gegen eine Lösung der Gleichung
gegen eine Lösung der Gleichung  ,
also gegen einen Fixpunkt
der Iterationsfunktion
,
also gegen einen Fixpunkt
der Iterationsfunktion  .
.
Ein systematisches Vorgehen bei der Suche nach geeigneten Algorithmen dieser
Gestalt ermöglicht die Idee der Splitting-Verfahren. Dabei wird die Matrix
in eine Summe

zerlegt mit einer leicht zu invertierenden Matrix
und dem Rest  .
Durch Einsetzen und Umstellen ergibt sich damit aus
die Fixpunktgleichung
.
Durch Einsetzen und Umstellen ergibt sich damit aus
die Fixpunktgleichung
 .
.
Mit  und
und  erhält man so ein Iterationsverfahren der Gestalt
erhält man so ein Iterationsverfahren der Gestalt  ,
das im Falle der Konvergenz die Lösung von
liefert. Die Konvergenzgeschwindigkeit ist umso größer, je kleiner der
betragsgrößte Eigenwert der Iterationsmatrix
ist. Dieser lässt sich auch durch beliebige Matrixnormen von
abschätzen.
,
das im Falle der Konvergenz die Lösung von
liefert. Die Konvergenzgeschwindigkeit ist umso größer, je kleiner der
betragsgrößte Eigenwert der Iterationsmatrix
ist. Dieser lässt sich auch durch beliebige Matrixnormen von
abschätzen.
Als klassische Beispiele für Splitting-Verfahren verwendet das
Jacobi-Verfahren für
die Diagonalmatrix mit der Hauptdiagonale von ,
das Gauß-Seidel-Verfahren
den unteren Dreiecksanteil von .
Zur Konvergenzbeschleunigung der Fixpunktverfahren lässt sich die Idee der Relaxation
nutzen. Denkt man sich die Iteration in der Form

mit der Korrektur  im -ten
Schritt dargestellt, geht man mit einem geeignet gewählten Relaxationsparameter
im -ten
Schritt dargestellt, geht man mit einem geeignet gewählten Relaxationsparameter
 zu
zu

über. Zum Beispiel erhält man auf diese Weise aus dem Gauß-Seidel-Verfahren das SOR-Verfahren.
Jacobi-Verfahren zur Eigenwertberechnung
Ein einfaches, aber zuverlässiges iteratives Verfahren zur Lösung des
Eigenwertproblems für symmetrische Matrizen ist das Jacobi-Verfahren.
Es erzeugt durch sukzessive Ähnlichkeitstransformationen mit Givens-Rotationen eine
Folge von symmetrischen Matrizen, die alle ähnlich zu der gegebenen
symmetrischen Matrix
sind und gegen eine Diagonalmatrix
konvergieren. Bricht man das Verfahren nach einer geeigneten Anzahl von
Schritten ab, erhält man deshalb mit den Diagonaleinträgen von
Näherungen für die gesuchten Eigenwerte von .
In jedem Schritt wird die Givens-Rotation, in diesem Zusammenhang auch als
Jacobi-Rotation bezeichnet, so gewählt, dass der Eintrag an der
Matrixposition  und der symmetrisch dazu liegende bei
und der symmetrisch dazu liegende bei  zu null transformiert werden. Dabei ist jedoch zu beachten, dass bei dieser
Transformation die ganze -te
und
zu null transformiert werden. Dabei ist jedoch zu beachten, dass bei dieser
Transformation die ganze -te
und  -te
Zeile sowie die ganze -te
und -te
Spalte der Matrix geändert wird. Deshalb werden die in einem Schritt erzeugten
Nullen im Allgemeinen in den folgenden Schritten wieder zunichtegemacht. Dennoch
konvergieren bei geeigneter Wahl der Positionen für die Jacobi-Rotationen alle
Nichtdiagonalelemente gegen null. Das klassische Jacobi-Verfahren wählt
dazu in jedem Iterationsschritt diejenige Position ,
an der sich das Nichtdiagonalelement mit dem größten Absolutbetrag befindet. Bei
einer Handrechnung war diese Position normalerweise schnell zu erkennen, bei der
Umsetzung als Computerprogramm ist der Aufwand für die Suche danach im Vergleich
zu den übrigen Rechenoperationen jedoch erheblich. Daher wird heute meist das
zyklische Jacobi-Verfahren verwendet. Dabei werden die Positionen in
einer vorher fest gewählten Reihenfolge zyklisch durchlaufen, etwa einfach
spaltenweise.
Es lässt sich beweisen, dass sowohl das klassische als auch das zyklische
Jacobi-Verfahren stets konvergieren. Im Vergleich zu moderneren Algorithmen ist
die Konvergenz allerdings relativ langsam. Für dünnbesetzte Matrizen ist das
Jacobi-Verfahren nicht geeignet, da im Laufe der Iteration die Matrix mit immer
mehr Nichtnulleinträgen aufgefüllt wird.
-te
Zeile sowie die ganze -te
und -te
Spalte der Matrix geändert wird. Deshalb werden die in einem Schritt erzeugten
Nullen im Allgemeinen in den folgenden Schritten wieder zunichtegemacht. Dennoch
konvergieren bei geeigneter Wahl der Positionen für die Jacobi-Rotationen alle
Nichtdiagonalelemente gegen null. Das klassische Jacobi-Verfahren wählt
dazu in jedem Iterationsschritt diejenige Position ,
an der sich das Nichtdiagonalelement mit dem größten Absolutbetrag befindet. Bei
einer Handrechnung war diese Position normalerweise schnell zu erkennen, bei der
Umsetzung als Computerprogramm ist der Aufwand für die Suche danach im Vergleich
zu den übrigen Rechenoperationen jedoch erheblich. Daher wird heute meist das
zyklische Jacobi-Verfahren verwendet. Dabei werden die Positionen in
einer vorher fest gewählten Reihenfolge zyklisch durchlaufen, etwa einfach
spaltenweise.
Es lässt sich beweisen, dass sowohl das klassische als auch das zyklische
Jacobi-Verfahren stets konvergieren. Im Vergleich zu moderneren Algorithmen ist
die Konvergenz allerdings relativ langsam. Für dünnbesetzte Matrizen ist das
Jacobi-Verfahren nicht geeignet, da im Laufe der Iteration die Matrix mit immer
mehr Nichtnulleinträgen aufgefüllt wird.
Vektoriteration
Eine einfache Ausgangsidee zur Berechnung von Eigenvektoren einer Matrix
ist die Potenzmethode.
Ein Startvektor  wird iterativ immer wieder mit
multipliziert
wird iterativ immer wieder mit
multipliziert

oder, ausgedrückt mit der -ten
Matrixpotenz, es wird  berechnet. Dahinter steckt die geometrische Anschauung, dass der Vektor durch
in jedem Schritt am stärksten in die Richtung des Eigenvektors mit dem größten
Eigenwert gestreckt wird. In dieser einfachen Form ist die Vektoriteration
jedoch für die Praxis ungeeignet, da im Allgemeinen die Einträge von
schnell sehr klein oder sehr groß werden. Daher wird der Vektor in jedem Schritt
zusätzlich zur Multiplikation mit
noch mit einer Vektornorm auf
berechnet. Dahinter steckt die geometrische Anschauung, dass der Vektor durch
in jedem Schritt am stärksten in die Richtung des Eigenvektors mit dem größten
Eigenwert gestreckt wird. In dieser einfachen Form ist die Vektoriteration
jedoch für die Praxis ungeeignet, da im Allgemeinen die Einträge von
schnell sehr klein oder sehr groß werden. Daher wird der Vektor in jedem Schritt
zusätzlich zur Multiplikation mit
noch mit einer Vektornorm auf  normiert. Man kann dann unter gewissen Voraussetzungen an die Lage der
Eigenwerte beweisen, dass dieses Verfahren bis auf möglicherweise einen skalaren
Vorfaktor tatsächlich gegen einen Eigenvektor zum betragsgrößten Eigenwert
konvergiert.
normiert. Man kann dann unter gewissen Voraussetzungen an die Lage der
Eigenwerte beweisen, dass dieses Verfahren bis auf möglicherweise einen skalaren
Vorfaktor tatsächlich gegen einen Eigenvektor zum betragsgrößten Eigenwert
konvergiert.
Wendet man diese Idee formal auf die inverse Matrix
an, so erhält man einen Eigenvektor zum betragskleinsten Eigenwert von
.
Hierzu wird freilich nicht die Inverse selbst berechnet, sondern es wird in
jedem Schritt das lineare Gleichungssystem

gelöst. Eine weitere Verallgemeinerung der Idee erhält man mithilfe eines
sogenannten Shiftparameters  .
Ein Eigenvektor von
zu dem am nächsten bei
liegenden Eigenwert ist nämlich auch ein Eigenvektor zum betragskleinsten
Eigenwert der „geshifteten“ Matrix
.
Ein Eigenvektor von
zu dem am nächsten bei
liegenden Eigenwert ist nämlich auch ein Eigenvektor zum betragskleinsten
Eigenwert der „geshifteten“ Matrix  .
Mit der zugehörigen Iteration
.
Mit der zugehörigen Iteration

und Normierung von
in jedem Schritt ergibt sich das Verfahren der
inversen
Vektoriteration.
Vektoriterationsverfahren berechnen also zunächst einen bestimmten
Eigenvektor von ,
der zugehörige Eigenwert kann mithilfe des Rayleigh-Quotienten
erhalten werden. Sie sind offenbar dann gut geeignet, wenn – wie häufig in
bestimmten Anwendungsfällen – nur der größte, nur der kleinste oder allgemeiner
nur ein einzelner Eigenwert mitsamt seinem Eigenvektor gesucht ist.
QR-Verfahren
Das QR-Verfahren ist zurzeit der wichtigste Algorithmus zu Berechnung aller
Eigenwerte und Eigenvektoren von nicht zu großen vollbesetzten Matrizen .
Es ist ein Iterationsverfahren, das in jedem Schritt eine QR-Zerlegung verwendet, um
durch wiederholte Ähnlichkeitstransformationen eine Matrixfolge zu erzeugen, die
schnell gegen eine obere Dreiecksmatrix konvergiert. Startend mit der
Ausgangsmatrix  wird in seiner Grundidee im -ten
Schritt die Matrix QR-zerlegt,
wird in seiner Grundidee im -ten
Schritt die Matrix QR-zerlegt,
 ,
,
und anschließend werden die beiden Faktoren in umgekehrter Reihenfolge wieder zusammenmultipliziert:
 ,
,
um die neue Näherungsmatrix zu erhalten. Wegen  ergibt sich
ergibt sich  und daraus
und daraus  ;
es handelt sich bei dieser Umformung also tatsächlich um eine
Ähnlichkeitstransformation mit einer orthogonalen Matrix. Wie eine genauere
Analyse zeigt, besteht ein enger Zusammenhang zur Potenzmethode: Die
QR-Iteration lässt sich auffassen als eine Potenzmethode, die simultan auf alle
Vektoren einer Orthonormalbasis angewendet wird; durch die QR-Zerlegung in jedem
Schritt wird dabei sichergestellt, dass diese Vektoren im Laufe der Iteration
auch numerisch stabil orthonormiert bleiben (siehe auch Unterraumiteration).
Aus dieser Darstellung ergibt sich auch ein Beweis, dass das Verfahren unter
geringen Voraussetzungen an
gegen eine obere Dreiecksmatrix konvergiert.
;
es handelt sich bei dieser Umformung also tatsächlich um eine
Ähnlichkeitstransformation mit einer orthogonalen Matrix. Wie eine genauere
Analyse zeigt, besteht ein enger Zusammenhang zur Potenzmethode: Die
QR-Iteration lässt sich auffassen als eine Potenzmethode, die simultan auf alle
Vektoren einer Orthonormalbasis angewendet wird; durch die QR-Zerlegung in jedem
Schritt wird dabei sichergestellt, dass diese Vektoren im Laufe der Iteration
auch numerisch stabil orthonormiert bleiben (siehe auch Unterraumiteration).
Aus dieser Darstellung ergibt sich auch ein Beweis, dass das Verfahren unter
geringen Voraussetzungen an
gegen eine obere Dreiecksmatrix konvergiert.
In dieser einfachen Form ist das QR-Verfahren aus zwei Gründen noch nicht für
die Praxis geeignet. Zum einen ist der Rechenaufwand für die QR-Zerlegung, die
in jedem Schritt bestimmt werden muss, sehr groß. Zum anderen findet die
Konvergenz im Allgemeinen nur langsam statt, es müssen also viele Schritte
durchgeführt werden, um eine gewünschte Genauigkeit zu erhalten. Dem ersten
Punkt lässt dich dadurch begegnen, dass in einem Vorbereitungsschritt die Matrix
durch Ähnlichkeitstransformationen auf
Hessenberg-Gestalt
gebracht wird. Das lässt sich durch
Transformationen mit geeigneten Householder-Matrizen erreichen. Da eine
Hessenberg-Matrix nur noch  Nichtnulleinträge unter der Hauptdiagonale hat, lässt sie sich schnell mit den
entsprechenden
Givens-Rotationen QR-zerlegen. Wie sich leicht zeigen lässt, erhält ein Schritt
des QR-Verfahrens Symmetrie und Hessenberg-Gestalt. Da eine symmetrische
Hessenberg-Matrix eine Tridiagonalmatrix ist, vereinfacht sich das Verfahren im
symmetrischen Fall nochmals erheblich. Die Konvergenzgeschwindigkeit kann
ähnlich wie bei der inversen Vektoriteration deutlich erhöht werden, wenn in
jedem Schritt anstelle der Matrix
Nichtnulleinträge unter der Hauptdiagonale hat, lässt sie sich schnell mit den
entsprechenden
Givens-Rotationen QR-zerlegen. Wie sich leicht zeigen lässt, erhält ein Schritt
des QR-Verfahrens Symmetrie und Hessenberg-Gestalt. Da eine symmetrische
Hessenberg-Matrix eine Tridiagonalmatrix ist, vereinfacht sich das Verfahren im
symmetrischen Fall nochmals erheblich. Die Konvergenzgeschwindigkeit kann
ähnlich wie bei der inversen Vektoriteration deutlich erhöht werden, wenn in
jedem Schritt anstelle der Matrix  die Matrix
die Matrix  mit einem geschickt gewählten Shiftparameter
mit einem geschickt gewählten Shiftparameter  transformiert wird. Für die Wahl von ,
der Wert sollte eine Näherung an einen Eigenwert von
sein, existieren verschiedene sogenannte Shiftstrategien.
transformiert wird. Für die Wahl von ,
der Wert sollte eine Näherung an einen Eigenwert von
sein, existieren verschiedene sogenannte Shiftstrategien.
Mit einer Variante des QR-Verfahrens kann auch die sogenannte Singulärwertzerlegung einer Matrix berechnet werden. Diese Verallgemeinerung der Diagonalisierung auf beliebige – sogar nicht quadratische – Matrizen wird in einigen Anwendungen, wie etwa in der Bildkompression, direkt verwendet. Mithilfe der Singulärwertzerlegung können auch große, schlecht konditionierte lineare Ausgleichsprobleme gelöst werden.
Krylow-Unterraum-Verfahren
Die Krylow-Unterraum-Verfahren mit ihren zahlreichen Varianten und
Spezialisierungen sind die wichtigste Verfahrensgruppe zur Lösung sowohl von
linearen Gleichungssystemen als auch von Eigenwertproblemen, wenn die gegebene
Matrix
sehr groß und dünnbesetzt ist. Der historisch erste Algorithmus aus dieser
Gruppe ist das Verfahren
der konjugierten Gradienten, kurz CG-Verfahren (von englisch conjugate
gradients) zur Lösung linearer Gleichungssysteme mit
symmetrischen und positiv definiten Koeffizientenmatrizen.
CG-Verfahren
Der fruchtbare Zusammenhang des CG-Verfahrens mit Krylow-Unterräumen
wurde erst später erkannt, seine Grundidee ist eine andere: Es löst anstelle des
Gleichungssystems ein dazu äquivalentes Optimierungsproblem.
Ist nämlich
symmetrisch und positiv definit, so ist die Lösung von
die eindeutig bestimmte Minimalstelle der Funktion
 .
.
Damit stehen grundsätzlich alle numerischen Verfahren zur Lösung von
Optimierungsproblemen auch für das lineare Gleichungssystem zur Verfügung,
insbesondere die sogenannten Abstiegsverfahren.
Das sind iterative Verfahren, die ausgehend von der aktuellen Näherung
im -ten
Schritt entlang einer geeigneten Suchrichtung  das eindimensionale Optimierungsproblem
das eindimensionale Optimierungsproblem
- „Suche
 ,
sodass
,
sodass  minimal wird.“
minimal wird.“
lösen. Die dabei gefundene Stelle  wird die neue Näherung für den nächsten Schritt. Eine zunächst naheliegende Wahl
für die Suchrichtung
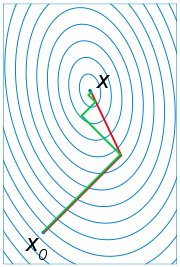
ist die Richtung des steilsten Abstiegs, was auf das Gradientenverfahren
zur Bestimmung der Minimalstelle führt. Allerdings zeigt sich, dass die so
berechneten Näherungen
sich im Allgemeinen nur sehr langsam und in einem „Zickzackkurs“ der wahren
Lösung annähern.
Wesentlich besser geeignet sind Suchrichtungen, die die spezielle Gestalt der zu
minimierenden Funktion
wird die neue Näherung für den nächsten Schritt. Eine zunächst naheliegende Wahl
für die Suchrichtung
ist die Richtung des steilsten Abstiegs, was auf das Gradientenverfahren
zur Bestimmung der Minimalstelle führt. Allerdings zeigt sich, dass die so
berechneten Näherungen
sich im Allgemeinen nur sehr langsam und in einem „Zickzackkurs“ der wahren
Lösung annähern.
Wesentlich besser geeignet sind Suchrichtungen, die die spezielle Gestalt der zu
minimierenden Funktion  berücksichtigen. Die Niveaumengen
von
sind -dimensionale
Ellipsoide (im anschaulichen,
zweidimensionalen Fall Ellipsen),
daher ist es günstig, die Suchrichtungen zueinander konjugiert zu wählen
(im Anschauungsfall entspricht das den konjugierten
Durchmessern). Dabei heißen zwei Richtungen
und
berücksichtigen. Die Niveaumengen
von
sind -dimensionale
Ellipsoide (im anschaulichen,
zweidimensionalen Fall Ellipsen),
daher ist es günstig, die Suchrichtungen zueinander konjugiert zu wählen
(im Anschauungsfall entspricht das den konjugierten
Durchmessern). Dabei heißen zwei Richtungen
und  konjugiert, wenn
konjugiert, wenn  gilt. Das CG-Verfahren wählt daher für die erste Suchrichtung die Richtung des
steilsten Abstiegs, aber die folgenden so, dass alle Suchrichtungen zueinander
konjugiert sind. Es lässt sich zeigen, dass dann nach
Abstiegen die wahre Lösung erreicht wird. Meist ist aber eine ausreichend genaue
Näherungslösung schon nach deutlich weniger Schritten erreicht und das Verfahren
kann vorzeitig abgebrochen werden.
gilt. Das CG-Verfahren wählt daher für die erste Suchrichtung die Richtung des
steilsten Abstiegs, aber die folgenden so, dass alle Suchrichtungen zueinander
konjugiert sind. Es lässt sich zeigen, dass dann nach
Abstiegen die wahre Lösung erreicht wird. Meist ist aber eine ausreichend genaue
Näherungslösung schon nach deutlich weniger Schritten erreicht und das Verfahren
kann vorzeitig abgebrochen werden.
Vom CG-Verfahren zu den Krylow-Unterraum-Verfahren
In den Rechenschritten des CG-Verfahrens geht die Matrix
nur in der Form von Matrix-Vektor-Produkten ein. Sie selbst wird nicht zerlegt
oder umgeformt – ein großer Vorteil, wenn sie dünnbesetzt ist. Nimmt man zur
Vereinfachung (aber ohne
Beschränkung der Allgemeinheit) an, dass als Startvektor
der Nullvektor gewählt wird, so zeigt eine genauere Analyse, dass jede Näherung
eine Linearkombination der Vektoren  ist, also aus wiederholten Multiplikationen der rechten Seite
mit
aufgebaut wird. Anders ausgedrückt: Jedes
liegt in einem Krylow-Unterraum
ist, also aus wiederholten Multiplikationen der rechten Seite
mit
aufgebaut wird. Anders ausgedrückt: Jedes
liegt in einem Krylow-Unterraum
 .
.
Diese Eigenschaft ist das Kennzeichen der Krylow-Unterraum-Verfahren: Sie
erzeugen iterativ für  Näherungen
mit
Näherungen
mit  .
Dabei wird
zusätzlich so gewählt, dass das Residuum
.
Dabei wird
zusätzlich so gewählt, dass das Residuum  in einem noch festzulegenden Sinne möglichst klein ist. Beim CG-Verfahren ist
die Bedingung nicht unbedingt naheliegend, aber für die spezielle Struktur des
Problems gut geeignet: Mit der durch
gewichteten Vektornorm
in einem noch festzulegenden Sinne möglichst klein ist. Beim CG-Verfahren ist
die Bedingung nicht unbedingt naheliegend, aber für die spezielle Struktur des
Problems gut geeignet: Mit der durch
gewichteten Vektornorm  ist in jedem Schritt
ist in jedem Schritt  minimal.
Der Nachteil liegt dabei darin, dass dies nur funktioniert, wenn
tatsächlich symmetrisch und positiv definit ist, anderenfalls ist
minimal.
Der Nachteil liegt dabei darin, dass dies nur funktioniert, wenn
tatsächlich symmetrisch und positiv definit ist, anderenfalls ist  gar keine Norm. Im Allgemeinen werden die Zusatzbedingungen, die
Krylow-Unterraum-Verfahren an die Wahl von
gar keine Norm. Im Allgemeinen werden die Zusatzbedingungen, die
Krylow-Unterraum-Verfahren an die Wahl von  stellen, als sogenannte Projektionsbedingung formuliert. Man verlangt dabei,
dass das Residuum
stellen, als sogenannte Projektionsbedingung formuliert. Man verlangt dabei,
dass das Residuum  orthogonal zu allen Vektoren aus einem -dimensionalen
Unterraum
orthogonal zu allen Vektoren aus einem -dimensionalen
Unterraum  ist, in Symbolen
ist, in Symbolen
 .
.
Die
sind normalerweise selbst Krylow-Unterräume, im einfachsten Fall, wie auch beim
CG-Verfahren, zum Beispiel  .
Für die konkrete Berechnung der Näherungen werden sukzessive Orthonormalbasen
der beteiligten Krylow-Unterräume aufgebaut. Das bekannte Gram-Schmidt-Verfahren
zur Orthonormalisierung ist in seiner Standardform leider numerisch instabil. Es
lässt sich jedoch mit einer kleinen Modifikation stabilisieren.
.
Für die konkrete Berechnung der Näherungen werden sukzessive Orthonormalbasen
der beteiligten Krylow-Unterräume aufgebaut. Das bekannte Gram-Schmidt-Verfahren
zur Orthonormalisierung ist in seiner Standardform leider numerisch instabil. Es
lässt sich jedoch mit einer kleinen Modifikation stabilisieren.
Weitere Krylow-Unterraum-Verfahren
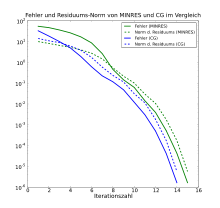
Aus den genannten Grundideen ergeben sich zahlreiche Variationen, Anpassungen
und Verbesserungen innerhalb dieser Verfahrensklasse, von denen nur einige
exemplarisch genannt werden sollen. Eine direkte Verallgemeinerung des
CG-Verfahrens ist das BiCG-Verfahren.
Es hebt die Einschränkung auf symmetrische Matrizen dadurch auf, dass es
zusätzlich zu dem mit
gebildeten Krylow-Unterräumen, auch die zur transponierten Matrix  gehörigen verwendet. Eine Optimierung, die die zusätzlichen Multiplikationen mit
vermeidet, ist das CGS-Verfahren.
Beide Verfahrenstypen sind in vielen praktischen Fällen instabil, bilden aber
die Grundlage für verschiedene Stabilisierungsversuche, etwa in der Gruppe der
BiCGSTAB-Verfahren.
Wichtige und im Allgemeinen stabile Verfahren sind GMRES und seine Spezialisierung
für symmetrische Matrizen, MINRES.
Sie setzen direkt bei den Residuen an und bestimmen
im Krylow-Unterraum so, dass
gehörigen verwendet. Eine Optimierung, die die zusätzlichen Multiplikationen mit
vermeidet, ist das CGS-Verfahren.
Beide Verfahrenstypen sind in vielen praktischen Fällen instabil, bilden aber
die Grundlage für verschiedene Stabilisierungsversuche, etwa in der Gruppe der
BiCGSTAB-Verfahren.
Wichtige und im Allgemeinen stabile Verfahren sind GMRES und seine Spezialisierung
für symmetrische Matrizen, MINRES.
Sie setzen direkt bei den Residuen an und bestimmen
im Krylow-Unterraum so, dass  minimal ist. Weitere Verbesserungen dieses Grundprinzips sind etwa das QMR-
und das TFQMR-Verfahren.
minimal ist. Weitere Verbesserungen dieses Grundprinzips sind etwa das QMR-
und das TFQMR-Verfahren.
Krylow-Unterraum-Verfahren können nicht nur für sehr große dünnbesetzte
lineare Gleichungssysteme verwendet werden, sondern auch zur Lösung ebensolcher
Eigenwertprobleme – ein weiterer Grund für ihre große Bedeutung in der modernen
numerischen linearen Algebra. Natürlich kann in Eigenwertproblemen nicht mit
 gestartet werden (
gestartet werden ( ist ja per Definition kein Eigenvektor). Es werden hier die Näherungen
und zugehörige
ist ja per Definition kein Eigenvektor). Es werden hier die Näherungen
und zugehörige  so bestimmt, dass
so bestimmt, dass

mit
gilt. Dieses Vorgehen führt auf ein nur -dimensionales
Eigenwertproblem, das sich für kleine
leicht lösen lässt und Näherungen an einige Eigenwerte von
liefert.
Der zugehörige Grundalgorithmus ist das Arnoldi-Verfahren.
Wie stets bei Eigenwertproblemen ergeben sich für symmetrische Matrizen
deutliche Vereinfachungen; diese führen auf das
Lanczos-Verfahren.
Literatur
- Steffen Börm, Christian Mehl: Numerical Methods for Eigenvalue Problems. Walter de Gruyter, Berlin/Boston 2012, ISBN 978-3-11-025033-6.
- Folkmar Bornemann: Numerische lineare Algebra – Eine konzise Einführung mit MATLAB und Julia. Springer Spektrum, Wiesbaden 2016, ISBN 978-3-658-12883-8.
- Biswa Nath Datta: Numerical Linear Algebra and Applications. 2. Auflage. SIAM, Philadelphia 2010, ISBN 978-0-89871-685-6.
- Andreas Meister: Numerik linearer Gleichungssysteme. 5. Auflage. Springer Spektrum, Wiesbaden 2015, ISBN 978-3-658-07199-8.
- Jean-Luc Chabert u.a. (Hrsg.): A History of Algorithms. Springer, Berlin/Heidelberg 1999, ISBN 978-3-540-63369-3.
- Gene H. Golub, Henk A. van der Vorst: Eigenvalue computation in the 20th century. In: Journal of Computational and Applied Mathematics. Band 123, 2000, S. 35–65.


© biancahoegel.de
Datum der letzten Änderung: Jena, den: 28.01. 2026