Haber-Bosch-Verfahren

Das Haber-Bosch-Verfahren ist ein großindustrielles chemisches Verfahren zur Synthese von Ammoniak. Es ist nach den deutschen Chemikern Fritz Haber und Carl Bosch benannt, die das Verfahren am Anfang des 20. Jahrhunderts entwickelten. Der zentrale Schritt des Verfahrens, die Ammoniaksynthese aus atmosphärischem Stickstoff und Wasserstoff, wird an einem eisenhaltigen Katalysator bei Drücken von etwa 150 bis 350 bar und Temperaturen von etwa 400 bis 500 °C durchgeführt. Als bedeutendes Chemieverfahren mit einem Produktionsausstoß von mehr als 150 Millionen Tonnen im Jahr 2017 deckt es mehr als 99 % der weltweiten Produktion an Ammoniak.
Ammoniak ist eine chemische Substanz, die überwiegend für die Herstellung von Harnstoff, Ammoniumnitrat, Ammoniumsulfat sowie Ammoniumphosphaten genutzt wird. Diese Stoffe werden als Düngemittel verwendet und tragen zur Ernährung eines Großteils der Weltbevölkerung bei. Weiterhin dient Ammoniak der Herstellung von Sprengstoffen und anderen stickstoffhaltigen Chemikalien. Die selektive katalytische Reduktion nutzt Ammoniak in der Rauchgasentstickung zur Umwandlung schädlicher Stickoxide in Stickstoff und Wasser. Ammoniak wird außerdem seit 1876 als umweltfreundliches und energieeffizientes Kältemittel eingesetzt.
Die wissenschaftlichen Leistungen zur Realisierung dieses Verfahrens waren zum einen die Untersuchung der zugrunde liegenden chemischen Reaktion durch Fritz Haber und Walther Nernst, zum anderen die systematische Suche nach geeigneten Katalysatoren durch Alwin Mittasch sowie die Klärung grundlegender verfahrenstechnischer Probleme für Hochdruckverfahren durch Carl Bosch. Im Zusammenhang mit dem Haber-Bosch-Verfahren vergab die Nobelstiftung 1918 den Nobelpreis für Chemie an Fritz Haber, 1931 an Carl Bosch sowie 2007 an Gerhard Ertl, der die vollständige theoretische Erklärung des Mechanismus der Ammoniakbildung fand.
Geschichte
Brot aus Luft

Seit Mitte des 19. Jahrhunderts ist durch die Arbeiten von Justus von Liebig bekannt, dass die Aufnahme von Stickstoffverbindungen eine Grundlage für das Wachstum von Nutzpflanzen ist. Dem Ackerboden wurden die notwendigen Stickstoffverbindungen über Mist, Kompost oder durch eine bestimmte Fruchtfolge zugeführt. Durch das rasante Anwachsen der Weltbevölkerung im 19. Jahrhundert konnte der damit einhergehende große Bedarf an Stickstoffdüngern nicht mehr durch natürliche Vorkommen von beispielsweise Guano oder Chilesalpeter und auch nicht durch technische Quellen wie Kokereigas gedeckt werden. Um auf diesen Umstand hinzuweisen, hielt der britische Chemiker William Crookes im Juni 1898 vor der British Association for the Advancement of Science in Bristol eine vielbeachtete Rede. Darin legte er dar, dass bis zum Jahr 1918 die Nachfrage nach Stickstoffverbindungen das Angebot bei weitem übersteigen werde und der westlichen Welt eine Hungersnot ungeahnten Ausmaßes drohe. Er führte weiterhin aus, dass die einzige Lösung dieses Problems die chemische Fixierung des in der Luft enthaltenen Stickstoffs sei. Die technische Umsetzung dieses Prozesses nannte er eine der großen Herausforderungen für die Chemiker seiner Zeit. Der unter dem Schlagwort „Brot aus Luft“ bekannte Versuch der Bindung des Luftstickstoffs in einer von Pflanzen aufnahmefähigen Chemikalie avancierte daraufhin zu einem der Schwerpunkte der chemischen Forschung der damaligen Zeit.
Frühe Entwicklungen

Ein erster Durchbruch bei der Fixierung des Luftstickstoffs gelang 1898 mit der Darstellung von Calciumcyanamid nach dem Frank-Caro-Verfahren, bei dem Calciumcarbid bei hohen Temperaturen atmosphärischen Stickstoff aufnimmt und so fixiert. Die Hydrolyse des Calciumcyanamids liefert Ammoniak und Calciumcarbonat. Obwohl große Kapazitäten für die Herstellung von Calciumcyanamid aufgebaut wurden, war das Verfahren auf Dauer wegen des hohen Energiebedarfs von etwa 190 Gigajoule pro Tonne Ammoniak nicht konkurrenzfähig.
Wilhelm Ostwald meldete im Jahre 1900 ein Patent zur „Herstellung von Ammoniak und Ammoniakverbindungen aus freiem Stickstoff und Wasserstoff“ an, da es ihm scheinbar gelungen war, Ammoniak katalytisch aus den Elementen herzustellen. Schon 1903 veröffentlichte der Rottweiler Sprengstoff-Hersteller Max Duttenhofer die Warnung Ostwalds vor einem Salpeter-Embargo im Kriegsfalle im Schwäbischen Merkur. Ostwald zog sein Patent zurück, nachdem Bosch nachgewiesen hatte, dass der entstandene Ammoniak aus dem Eisennitrid des verwendeten Katalysators stammte.
Das Birkeland-Eyde-Verfahren, das von dem norwegischen Wissenschaftler Kristian Birkeland und seinem Geschäftspartner Sam Eyde entwickelt und 1903 in Betrieb genommen wurde, fixierte den Luftstickstoff, indem er ihn mittels eines elektrischen Lichtbogens direkt zu Stickstoffmonoxid oxidierte. Beim Abkühlen und weiterer Oxidation bildete sich Distickstoffpentoxid, das durch Absorption in Wasser zu Salpetersäure reagierte. Die geringe Energieeffizienz führte schon bald zur Verdrängung des Verfahrens.
Technische Realisierung
.jpg)
Die Bereitstellung vor allem des Rohstoffs Wasserstoff, der in größeren Mengen bis dahin nur bei der Chloralkali-Elektrolyse anfiel, erforderte völlig neue Prozesse. Auch für den Bau der zur Ammoniaksynthese benötigten chemischen Reaktoren, in denen Wasserstoff bei hohen Drücken und Temperaturen kontrolliert werden konnte, gab es bis dahin keine Referenzen in der Technik. Carl Bosch und Fritz Haber entwickelten daraufhin neue Lösungen in vielen Bereichen der technischen Chemie und des Reaktorbaus.
Aufgrund der großen Anzahl an benötigten Spezialisten zur Umsetzung der Herstellung im industriellen Maßstab gründete Bosch einen interdisziplinären Arbeitsbereich Chemietechnik, in dem Maschinenbauingenieure und Chemiker zusammen arbeiteten. Da die zunächst für den Reaktorbau verwendeten Stähle durch atomar eindiffundierten Wasserstoff erodierten, war eine Aufgabe des neuen Arbeitsbereiches die Erforschung von Werkstoffschäden durch Entkohlung der Kohlenstoffstähle. Dies führte schließlich zur Entwicklung hochlegierter Chrom-Nickel-Stähle, die einem Wasserstoffangriff bei den benötigten Reaktionstemperaturen und -drücken standhielten. Insbesondere das von Julius Schierenbeck entwickelte Schierenbeck-Wickelverfahren, bei dem auf ein chemisch widerstandsfähiges Zentralrohr mehrere Lagen eines heißen Metallbandes aufgeschrumpft wurden, ermöglichte den Bau größerer und sicherer Hochdruckreaktoren. Alwin Mittasch entwickelte und testete parallel dazu in 20.000 Versuchen etwa 3000 verschiedene Katalysatoren auf Basis von Eisenoxid mit verschiedenen umsetzungsbeschleunigenden Substanzen, die er Aktivator oder Promotor nannte. Der im Jahr 2015 verwendete Katalysator entsprach noch weitgehend dem von Mittasch entwickelten.

Im Jahr 1913 nahm die BASF erstmals eine Anlage nach dem Haber-Bosch-Verfahren im Werk Ludwigshafen-Oppau in Betrieb. Die Kapazität der Anlage betrug anfangs 30 Tonnen pro Tag. Bereits 1914 wurde die Weiterentwicklung des Verfahrens bis zur großindustriellen Anwendbarkeit durch den deutschen Generalstabschef Erich von Falkenhayn forciert, woraufhin Bosch das sogenannte Salpeterversprechen abgab. Dabei handelte es sich um einen Vertrag zur Lieferung von Nitraten mit staatlichen Abnahmegarantien und unter finanzieller Unterstützung durch das Reich für den Bau entsprechender Anlagen. Damit sollte Ammoniumnitrat als Grundlage militärischer Sprengstoffe in ausreichendem Maße ohne den sonst verwendeten natürlich vorkommenden Salpeter produziert werden können. Kurz darauf gelang es durch das Haber-Bosch-Verfahren, entsprechend große Mengen des kriegswichtigen Materials herzustellen. Damit konnte das durch die britische Seeblockade von Stickstoffquellen wie Chilesalpeter abgeschnittene Deutsche Reich im Ersten Weltkrieg seine Munitions- und auch seine Düngemittelproduktion aufrechterhalten und den wirtschaftlichen Zusammenbruch abwenden. Neben der Großanlage in Oppau bei Ludwigshafen wurden weitere in Leuna und Bitterfeld durch die BASF und nach Fusion im deutschen Großkonzern durch die I.G. Farben betrieben.
Nach dem Ersten Weltkrieg

Nach dem Krieg verpflichtete die Siegermacht Frankreich die BASF durch ein Abkommen, sämtliche Patente und Erfahrungen des Verfahrens herauszugeben und den Aufbau einer entsprechenden Fabrik in Toulouse zu unterstützen. Weitere Ammoniakanlagen entstanden zur selben Zeit in England, Italien und anderen Ländern. Diesen Bauvorhaben lag entweder eine Lizenz der BASF oder eine Verfahrensvariante mit veränderten Prozessparametern zugrunde, wozu das Casale-Verfahren und das Mont-Cenis-Uhde-Verfahren von dem 1921 gegründeten Ingenieurbüro Friedrich Uhde mit modifiziertem Katalysator zu zählen sind.
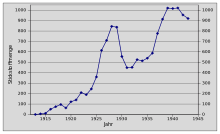
In die Zeit zwischen den Weltkriegen fiel die Gründung der I.G. Farben mit Carl Bosch als ihrem ersten Vorstandsvorsitzenden. Bereits 1926 betrug die Marktkapitalisierung des Unternehmens mit seinen mittlerweile 100.000 Mitarbeitern rund 1,4 Milliarden Reichsmark. Durch die mit dem New Yorker Börsencrash vom Oktober 1929 beginnende Weltwirtschaftskrise verringerte sich die Nachfrage nach synthetischem Ammoniak erheblich. Die Produktion in Deutschland, die bereits ein Jahresvolumen von über 800.000 Tonnen erreicht hatte, sank daraufhin auf unter 500.000 Tonnen und die Einnahmen der I.G. Farben halbierten sich. Dennoch blieb die I.G. Farben bis 1930 der weltweit größte Hersteller von Ammoniak; 65 Prozent des Gesamtgewinns entfielen auf die Ammoniaksynthese.
Das vom Kabinett Brüning erlassene Einfuhrembargo für Stickstoffdünger erlaubte der I.G. Farben, die Preise für synthetische Düngemittel wieder zu erhöhen. Nach der Machtergreifung Hitlers übernahm das Nazi-Regime die Kontrolle über die I.G. Farben. Bosch stellte auf Druck des NS-Regimes 1935 seinen Vorstandsposten zur Verfügung, der an das NSDAP-Mitglied und Wehrwirtschaftsführer Hermann Schmitz fiel. 1940 erreichte die Ammoniakproduktion in Deutschland bereits eine Million Tonnen pro Jahr. Infolge des stetig steigenden Bedarfs an Ammoniak und seinen Folgeprodukten entstanden immer leistungsfähigere Reaktoren.
Nach dem Zweiten Weltkrieg
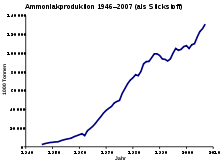
produktion von 1946 bis 2007
Die zunehmende Verfügbarkeit von preiswertem Erdöl und kostenreduzierenden Vergasungsprozessen durch beispielsweise die partielle Oxidation von Erdölfraktionen ermöglichte nach dem Zweiten Weltkrieg den Aufbau von Haber-Bosch-Anlagen in aller Welt. Die ursprünglich von der I.G. Farben entwickelte partielle Oxidation wurde durch das Unternehmen Imperial Chemical Industries (ICI) verbessert und um die Oxidation von Naphtha erweitert, womit in den 1950er und 1960er Jahren die Rohstoffe des Verfahrens preiswerter wurden.
Engineering-Unternehmen wie M. W. Kellogg entwickelten in der Folgezeit energieeffizientere und damit kostengünstigere Großanlagen mit nur einem Reaktor, die zu einer weltweiten Erhöhung der Anlagenkapazität führten. Zunehmender Wettbewerb und ein hoher Kostendruck ebneten schließlich den Weg für das LCA-Verfahren der ICI und das KAAP-Verfahren von Kellogg, Brown & Root, bei dem erstmals ein Rutheniumkatalysator Verwendung fand.
Rohstoffe
.jpg)
Ammoniak entsteht in einer Gleichgewichtsreaktion aus den Elementen Wasserstoff und Stickstoff gemäß der Gleichung
 ,
,
wobei der benötigte Stickstoff der Umgebungsluft entnommen wird. Der ebenfalls in der Luft enthaltene, aber unerwünschte Sauerstoff wurde zunächst durch Reduktion mit Wasserstoff in Wasser umgesetzt und so abgeschieden; das Fauser-Verfahren nutzte den bei der Ammoniakverbrennung mit Luft nicht umgesetzten Stickstoff als Rohstoff. Später erwies sich die Stickstoffgewinnung durch Luftzerlegung nach dem Linde-Verfahren als wirtschaftlicher.
Den größten Teil der Produktionskosten verursacht die Beschaffung von Wasserstoff, der zunächst aus der kostenintensiven Chloralkali-Elektrolyse gewonnen wurde. Mit Erdgas, Erdöl, Kohle sowie den Elektrolyseprodukten von Wasser kamen später weitere Quellen zur Erzeugung von Wasserstoff hinzu.
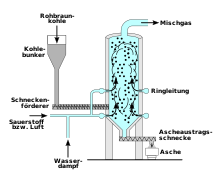
Die BASF verwendete Wassergas auf Basis der Kohlevergasung von Braunkohle mittels eines Winkler-Generators als primäre Quelle. Der Wasserstoff wird dabei über die Reaktion von Wasserdampf mit glühendem Koks gewonnen. Die zugeführte Luft wird derart dosiert, dass der Sauerstoff vollständig zu Kohlenstoffmonoxid umgesetzt wird. Der für die spätere Ammoniaksynthese erforderliche Stickstoff verblieb im Wassergas. Anschließend wurde das Kohlenstoffmonoxid mittels Konvertierung in leicht zu entfernendes Kohlenstoffdioxid oder in einer Wassergas-Shift-Reaktion zur Bereitstellung weiteren Wasserstoffs verwendet. Mit dem Rohstoff Kohle wurden im Jahr 2008 etwa 18 Prozent des weltweit produzierten Wasserstoffs hergestellt.
Obwohl Erdgas in den 1920er Jahren der BASF noch nicht als Rohstoff für die Herstellung von Wasserstoff zur Verfügung stand, initiierte Carl Bosch bereits die Entwicklung der Dampfreformierung von Methan, welche später ein wichtiger Bestandteil des Verfahrens werden sollte. Einen Durchbruch erzielte Georg Schiller für die I.G. Farben, dem die Dampfreformierung von Methan mittels eines Nickeloxid-Katalysators gelang. Der Standard Oil of New Jersey wurde eine Lizenz erteilt, die 1931 mit der Wasserstoffproduktion durch Dampfreformierung in ihrem Werk in Baton Rouge in Louisiana begann. Auf die Dampfreformierung von Erdgas entfielen 2014 etwa 48 Prozent der globalen Wasserstoffproduktion, zirka 60 Prozent davon verwendete das Haber-Bosch-Verfahren.
Ein anderes mögliches Verfahren der Wasserstoffgewinnung ist die partielle Oxidation Dabei werden erdgas- oder erdölstämmige Kohlenwasserstoffe mit Sauerstoff und Wasserdampf in einem offenen Reaktor ohne Katalysator bei etwa 1100 °C vergast und das Synthesegas wie bei der Dampfreformierung weiterverarbeitet. Die höheren Kohlenwasserstoffe aus Erdöl trugen 2008 zu 30 Prozent zur jährlichen Produktion von Wasserstoff bei.
Wasserstoff kann weiterhin durch die Elektrolyse von Wasser gewonnen werden. Hierdurch wird mit elektrischer Energie Wasser in Wasserstoff (H2) und Sauerstoff (O2) zerlegt. Dieses Verfahren ist nur wirtschaftlich, wenn preiswerte elektrische Energie zum Beispiel aus Wasserkraft zur Verfügung steht. Auf die Elektrolyse entfielen 2008 etwa vier Prozent der Wasserstoffproduktion. Angesichts der Klimaproblematik und dem Bestreben, CO2-Emissionen zu verringern, ist diese Möglichkeit der Wasserstoffgewinnung unter Verwendung erneuerbarer Energien wie Windkraft oder Solarstrom wieder stark in den Fokus der Politik und Wirtschaft gelangt. Thyssenkrupp Uhde Chlorine Engineers hat in den letzten Jahren seine Fertigungskapazitäten für die Alkalische Wasserelektrolyse auf 1 Gigawatt Elektrolyseurleistung pro Jahr ausgebaut.
Mit dem Aufkommen von Platforming-Kapazitäten in den Vereinigten Staaten Mitte der 1950er Jahre stand eine petrochemische Wasserstoffquelle zur Verfügung, die 1956 etwa elf Prozent des benötigten Wasserstoffs für die Ammoniaksynthese in den Vereinigten Staaten bereitstellte. Später nutzten andere Raffinerieverfahren wie die Hydrodesulfurierung sowie das Hydrocracken den anfallenden Wasserstoff.
Katalysator
.jpg)
Zur Senkung der Aktivierungsenergie und der damit einhergehenden Steigerung der Reaktionsgeschwindigkeit wird bei vielen chemischen Prozessen ein zusätzlicher Stoff, der Katalysator, eingesetzt, der während der Reaktion nicht verbraucht wird. Unterscheidet sich dabei der Aggregatzustand des Katalysators von dem der eigentlichen miteinander reagierenden Stoffe, handelt es sich um einen heterogenen Katalysator. So auch beim Haber-Bosch-Verfahren, bei dem feinverteiltes Eisen auf einem Eisenoxidträger in fester Form als Katalysator innerhalb eines reagierenden Gasgemisches dient. Dieser heterogene Katalysator, auch „Kontakt“ genannt, entsteht während der Reaktion aus einem anderen, zuvor im Reaktor eingebrachten Material, dem „Katalysatorvorläufer“ oder „Präkursorkontakt“.
Eisenkatalysator
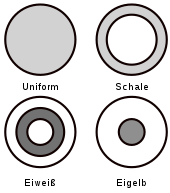
Der heterogene Eisenkatalysator ist eine katalytisch sehr aktive Form des kubisch raumzentrierten α-Eisens und entsteht durch chemische Reduktion aus einer speziellen Form oxidierten Eisens, dem Magnetit (Fe3O4). Die Wirkung des Katalysators wird durch oxidische Promotoren verstärkt, die dem Magnetit zuvor zugesetzt wurden. Im Falle der Ammoniaksynthese zählen dazu beispielsweise Aluminiumoxid, Kaliumoxid, Calciumoxid und Magnesiumoxid.
Die Herstellung des benötigten Magnetitkontakts erfordert einen speziellen Schmelzprozess, bei dem die verwendeten Rohmaterialien frei von Katalysatorgiften und die Promotorenzuschläge gleichmäßig in der Magnetit-Schmelze verteilt sein müssen. Durch schnelles Abkühlen der etwa 3500 °C heißen Magnetit-Schmelze bildet sich der gewünschte Katalysator mit hoher Aktivität, wodurch die Abriebresistenz desselben vermindert wird. Trotz dieses Nachteils wird in der Praxis die Methode des schnellen Abkühlens häufig bevorzugt.
Die Reduktion des Katalysatorvorläufers Magnetit zu α-Eisen wird mit Synthesegas direkt in der Produktionsanlage durchgeführt. Die Reduktion des Magnetits verläuft zunächst über die Stufe von Wüstit (FeO), sodass sich ein Kontakt mit einem Kern aus Magnetit bildet, der von einer Hülle aus Wüstit umgeben ist. Die weitere Reduktion der Magnetit- und Wüstit-Phase führt zur Bildung von α-Eisen, das zusammen mit den Promotoren die äußere Schale bildet. Die dabei ablaufenden Prozesse sind komplex und hängen von der Reduktionstemperatur ab. So disproportioniert Wüstit bei tieferen Temperaturen in eine Eisen- und eine Magnetitphase, bei höheren Temperaturen ist die Reduktion der Wüstit- und Magnetitphase zum Eisen der dominante Prozess.
Das α-Eisen bildet Primärkristallite mit einem Durchmesser von etwa 30 Nanometern. Diese bilden ein bimodales Porensystem mit Porendurchmessern von etwa 10 Nanometern, die durch Reduktion der Magnetit-Phase entstehen, beziehungsweise von 25 bis 50 Nanometer, die durch Reduktion der Wüstit-Phase entstehen. Die Promotoren werden mit Ausnahme von Cobaltoxid nicht reduziert.
Bei der Reduktion des Eisenoxids mit Synthesegas entsteht als Nebenprodukt Wasserdampf. Für eine optimale Katalysatorqualität muss dieser Wasserdampf berücksichtigt werden. Wenn dieser in Kontakt mit dem fein verteilten Eisen kommt, führt das speziell in Verbindung mit hohen Temperaturen zu vorzeitiger Alterung des Katalysators durch Rekristallisation. Daher wird der Dampfdruck des bei der Katalysatorbildung entstehenden Wassers im Gasgemisch möglichst gering gehalten, wobei Werte von unter 3 gm−3 angestrebt werden. Die Reduktion wird aus diesem Grund bei hohem Gasaustausch, geringem Druck und niedrigen Temperaturen durchgeführt. Die Exothermie der Ammoniakbildung sorgt für eine schrittweise Erhöhung der Temperatur.
Die Reduktion von frischem, vollständig oxidiertem Katalysator beziehungsweise Präkursor bis hin zum Erreichen der vollen Kapazität dauert vier bis zehn Tage. Die Wüstit-Phase wird schneller als die Magnetit-Phase (Fe3O4) und bei geringeren Temperaturen reduziert. Nach detaillierten kinetischen, mikroskopischen und röntgenspektroskopischen Untersuchungen konnte nachgewiesen werden, dass sich Wüstit als Erstes zu metallischem Eisen umsetzt. Dies führt zu einer Dichteinhomogenität (Gradient) der Eisen(II)-ionen, wodurch diese vom Magnetit durch das Wüstit an die Grenzfläche diffundieren und dort als Eisenkeime ausfallen.
In der technischen Praxis haben vorreduzierte, stabilisierte Katalysatoren einen bedeutenden Marktanteil errungen. Sie verfügen bereits über die voll ausgebildete Porenstruktur, sind jedoch nach der Herstellung wieder an der Oberfläche oxidiert worden und damit nicht mehr pyrophor. Das Reaktivieren solcher vorreduzierten Katalysatoren benötigt lediglich 30 bis 40 Stunden anstelle sonst üblicher mehrtägiger Zeitspannen. Neben der geringen Anlaufzeit besitzen sie mit einer höheren Wasserresistenz und einem geringeren Gewicht weitere Vorteile.
| Zusammensetzung eines Kontakts | % Eisen | % Kalium | % Aluminium | % Calcium | % Sauerstoff |
|---|---|---|---|---|---|
| Volumenzusammensetzung | 40,5 | 0,35 | 2,0 | 1,7 | 53,2 |
| Oberflächenzusammensetzung vor Reduktion | 8,6 | 36,1 | 10,7 | 4,7 | 40,0 |
| Oberflächenzusammensetzung nach Reduktion | 11,0 | 27,0 | 17,0 | 4,0 | 41,0 |
Andere Katalysatoren als Eisen
Seit der industriellen Einführung des Haber-Bosch-Verfahrens wurden viele Anstrengungen zu dessen Verbesserung unternommen, in deren Folge es zu bedeutenden Fortschritten kam. Bei der Verbesserung des Katalysators zur Ammoniaksynthese hingegen gab es seit den 1920ern lange Zeit keine nennenswerten Fortschritte.
Im Rahmen der Suche nach geeigneten Katalysatoren wurden viele Metalle intensiv getestet: Die Voraussetzung zur Eignung ist die dissoziativ verlaufende Adsorption des Stickstoffs (das Stickstoff-Molekül muss also während der Adsorption in zwei Stickstoff-Atome gespalten werden). Gleichzeitig darf die Bindung der Stickstoff-Atome nicht zu stark erfolgen, anderweitig käme es zur Herabsetzung der katalytischen Fähigkeiten (also zur Selbstvergiftung). Die Metalle im Periodensystem der Elemente links der Eisengruppe zeigen eine solche zu starke Bindung zu Stickstoff. Durch die damit verbundene Bildung von Volumen- oder Oberflächennitriden werden beispielsweise Chrom-Katalysatoren unwirksam, sie vergiften sich selbst. Metalle rechts der Eisengruppe hingegen adsorbieren Stickstoff in zu geringem Maße, als dass sie ausreichend Stickstoff für die Ammoniaksynthese zu aktivieren in der Lage wären. Haber selbst verwendete zunächst Osmium und Uran als Katalysatoren. Uran reagiert während der Katalyse zum Nitrid und Osmiumoxid ist sehr selten, flüchtig und hochgiftig.
Aufgrund des vergleichsweise geringen Preises, der großen Verfügbarkeit, der einfachen Verarbeitung, der Lebensspanne und der Aktivität wurde schließlich Eisen als Katalysator gewählt. Für eine Produktionskapazität von beispielsweise 1800 Tonnen pro Tag wird damit ein Druck von mindestens 130 bar, Temperaturen von 400 bis 500 °C und ein Reaktorvolumen von wenigstens 100 m³ benötigt. Theoretischen und praktischen Untersuchungen zufolge ist der Spielraum für weitere Verbesserungen des reinen Eisenkatalysators begrenzt. Erst die seit 1984 eingesetzte Modifizierung des Eisenkatalysators durch Cobalt steigerte dessen Aktivität merklich.
Katalysatoren der zweiten Generation
Auf Ruthenium basierende Katalysatoren zeigen bei vergleichbaren Drücken und niedrigeren Temperaturen eine höhere Aktivität und werden daher als Katalysatoren der zweiten Generation bezeichnet. Ihre Aktivität ist stark vom Katalysatorträger und den Promotoren abhängig. Als Träger kommen eine Vielzahl von Substanzen infrage, neben Kohlenstoff sind dies Magnesiumoxid, Aluminiumoxid, Zeolite, Spinelle und Bornitrid.
Ruthenium-Aktivkohle-Katalysatoren werden seit 1992 industriell im „KBR Advanced Ammonia Process“ (KAAP, dt. etwa weiterentwickelter Ammoniak-Prozess nach Kellogg, Brown und Root) verwendet. Der Kohlenstoffträger wird teilweise zu Methan abgebaut, was durch eine spezielle Behandlung des Kohlenstoffs bei 1500 °C abgemildert werden kann und so die Lebenszeit zu verlängern hilft. Daneben geht von dem fein dispergierten Kohlenstoff eine Explosionsgefahr aus. Aus diesen Gründen sowie aufgrund der niedrigen Acidität hat sich Magnesiumoxid als gute Alternative erwiesen. Träger mit aciden Eigenschaften entziehen dem Ruthenium Elektronen, machen es weniger reaktiv, und sie binden unerwünschterweise Ammoniak an der Oberfläche.
Katalysatorgifte
Katalysatorgifte reduzieren die Aktivität des Katalysators. Sie sind entweder Bestandteil des Synthesegases oder stammen aus Verunreinigungen des Katalysators selbst, wobei Letzteres keine größere Rolle spielt. Wasser, Kohlenstoffmonoxid, Kohlenstoffdioxid und Sauerstoff sind temporäre Katalysatorgifte. Schwefel-, Phosphor-, Arsen- und Chlor-Verbindungen sind permanente Katalysatorgifte.
Chemisch inerte Bestandteile des Synthesegasgemischs wie Edelgase oder Methan sind zwar keine Katalysatorgifte im eigentlichen Sinn, sie reichern sich aber durch die Zyklisierung der Prozessgase an und reduzieren so den Partialdruck der Reaktanden, was wiederum negativ auf die katalytische Umsetzung wirkt.
Reaktionstechnik
Synthesebedingungen
| Temperatur (°C) | Keq |
|---|---|
| 300 | 4,34 × 10−3 |
| 400 | 1,64 × 10−4 |
| 450 | 4,51 × 10−5 |
| 500 | 1,45 × 10−5 |
| 550 | 5,38 × 10−6 |
| 600 | 2,25 × 10−6 |
Die Ammoniaksynthese findet bei einem Mengenverhältnis Stickstoff zu Wasserstoff von 1 zu 3, einem Druck von 250 bis 350 bar, einer Temperatur von 450 bis 550 °C und unter Verwendung von α-Eisen als Katalysator gemäß folgender Gleichung statt:

Die Reaktion ist eine exotherme, unter Volumenverminderung ablaufende Gleichgewichtsreaktion, deren Massenwirkungskonstante Keq sich aus folgender Gleichung ergibt:
 .
.
Da die Reaktion exotherm ist, verschiebt sich das Gleichgewicht der Reaktion bei niedrigeren Temperaturen auf die Seite des Ammoniaks. Weiterhin entstehen aus vier Volumenteilen der Rohmaterialien zwei Volumenteile von Ammoniak. Gemäß dem Prinzip vom kleinsten Zwang begünstigt ein hoher Druck daher ebenfalls die Entstehung von Ammoniak. Es ist zudem ein hoher Druck notwendig, um eine ausreichende Oberflächenbedeckung des Katalysators mit Stickstoff zu gewährleisten.
Der Katalysator Ferrit (α-Fe) entsteht im Reaktor durch die Reduktion von Magnetit mit Wasserstoff. Dieser ist ab Temperaturen von etwa 400 bis 500 °C optimal wirksam. Durch den Katalysator wird die Aktivierungsbarriere für die Spaltung der Dreifachbindung des Stickstoffmoleküls stark abgesenkt, dennoch sind hohe Temperaturen für eine angemessene Reaktionsgeschwindigkeit erforderlich. Bei der gewählten Reaktionstemperatur liegt das Optimum zwischen dem Zerfall von Ammoniak in die Ausgangsstoffe und der Wirksamkeit des Katalysators. Das gebildete Ammoniak wird laufend aus dem Reaktionssystem entfernt. Der Volumenanteil von Ammoniak im Gasgemisch beträgt rund 20 %.
Die inerten Bestandteile, besonders die Edelgase wie Argon, dürfen einen bestimmten Gehalt nicht überschreiten, um den Partialdruck der Reaktanden nicht zu sehr abzusenken. Zur Entfernung der inerten Gasbestandteile wird ein Teil des Gases abgezogen und das Argon in einer Gastrennanlage abgeschieden. Die Gewinnung reinen Argons aus dem Kreislaufgas ist mittels Linde-Verfahren möglich.
Großtechnische Durchführung
Moderne Ammoniakanlagen erzeugen mehr als 3000 Tonnen pro Tag in einer Produktionslinie. Das folgende Schema zeigt den Aufbau einer Haber-Bosch-Anlage.
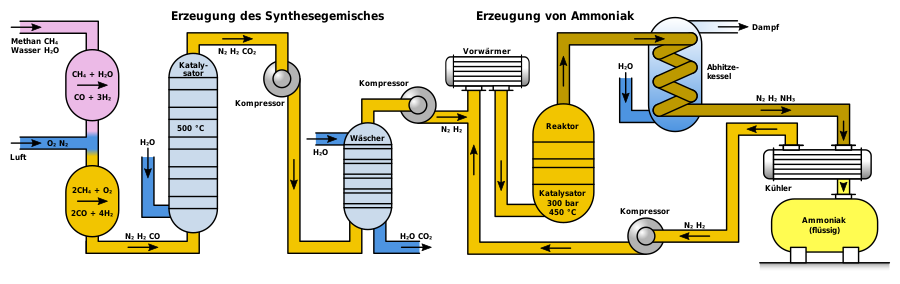
Je nach Herkunft des Synthesegases muss dieses zunächst von Verunreinigungen wie Schwefelwasserstoff oder organischen Schwefelverbindungen befreit werden, die als Katalysatorgift wirken. Hohe Konzentrationen von Schwefelwasserstoff, die bei Synthesegas aus Schwelkoksen vorkommen, werden in einer Nassreinigungsstufe wie dem Sulfosolvan-Verfahren entfernt, niedrige Konzentrationen durch Adsorption an Aktivkohle. Organoschwefelverbindungen werden mittels Druckwechseladsorption zusammen mit Kohlenstoffdioxid nach der CO-Konvertierung abgeschieden.
Zur Herstellung von Wasserstoff mittels Dampfreformierung reagiert Methan mit Wasserdampf mit Hilfe eines Nickeloxid-Aluminiumoxid-Katalysators im Primärreformer zu Kohlenstoffmonoxid und Wasserstoff. Die dafür benötigte Energie, die Enthalpie ΔH, beträgt dabei 206 kJ/mol.

Im Primärreformer setzt sich das Methangas nur unvollkommen um. Um die Ausbeute an Wasserstoff zu erhöhen und den Gehalt an inerten Bestandteilen so gering wie möglich zu halten, wird in einem zweiten Schritt im Sekundärreformer das restliche Methangas mit Sauerstoff zu Kohlenstoffmonoxid und Wasserstoff umgesetzt. Der Sekundärreformer wird hierzu mit Luft beschickt, wobei auch der für die spätere Ammoniaksynthese erforderliche Stickstoff in das Gasgemisch kommt.

In einem dritten Schritt wird das Kohlenstoffmonoxid zu Kohlenstoffdioxid oxidiert, was als CO-Konvertierung oder Wassergas-Shift-Reaktion bezeichnet wird.

Kohlenstoffmonoxid und Kohlenstoffdioxid bilden mit Ammoniak Carbamate, die als Feststoffe in kurzer Zeit Rohrleitungen und Apparate verstopfen würden. Im folgenden Prozessschritt muss daher das Kohlenstoffdioxid aus dem Gasgemisch entfernt werden. Im Gegensatz zu Kohlenstoffmonoxid kann Kohlenstoffdioxid durch eine Gaswäsche mit Triethanolamin leicht aus dem Gasgemisch entfernt werden. Das Gasgemisch enthält dann noch Edelgase wie Argon sowie Methan, die sich inert verhalten.
Anschließend wird das Gasgemisch mittels Turbokompressoren auf den benötigten Betriebsdruck komprimiert. Die entstehende Verdichtungswärme wird mittels Wärmetauschern abgeführt; sie wird zur Vorheizung von Rohgasen eingesetzt.
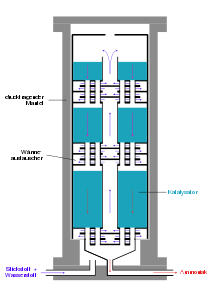
Im Ammoniakreaktor findet die eigentliche Herstellung von Ammoniak statt, wobei die ersten Reaktoren unter dem hohen Druck platzten, da der atomare Wasserstoff im kohlenstoffhaltigen Stahl zu Methan teilrekombinierte und Risse im Stahl erzeugte. Deshalb entwickelte Bosch Rohrreaktoren, bestehend aus einem drucktragenden Stahlrohr, in dem ein Futterrohr aus kohlenstoffarmem Eisen einzogen wurde, in welches der Katalysator eingefüllt wurde. Durch das innere Stahlrohr diffundierender Wasserstoff entwich nach außen über dünne Bohrungen des äußeren Stahlmantels, den sogenannten Bosch-Löchern. Die Entwicklung wasserstoffbeständiger Chrom-Molybdän-Stähle erlaubte die Konstruktion einwandiger Rohre. Ein Nachteil der Rohrreaktoren war der relativ hohe Druckverlust, der durch Kompression wieder aufgebracht werden musste.
Moderne Ammoniak-Reaktoren sind als Etagenreaktoren mit geringem Druckverlust ausgeführt, bei denen die Kontakte als Schüttungen auf etwa zehn übereinander befindlichen Etagen verteilt sind. Sie werden von oben nach unten nacheinander vom Gasgemisch durchströmt. Zur Kühlung wird seitlich Kaltgas eingedüst. Ein Nachteil dieses Reaktortyps ist die unvollständige Umsetzung des Kaltgasgemischs im letzten Katalysatorbett.
Alternativ wird zwischen den Katalysatorschichten das Reaktionsgemisch mittels Wärmetauschern gekühlt, wobei das Wasserstoff-Stickstoff-Gemisch auf Reaktionstemperatur vorgeheizt wird. Reaktoren dieses Typs weisen drei Katalysatorbetten auf. Neben einer guten Temperaturkontrolle besteht bei diesem Reaktortyp der Vorteil einer besseren Umsetzung der Rohstoffgase gegenüber Reaktoren mit Kaltgaseinspeisung. Im Uhde-Reaktor werden die Katalysatorbetten sogar radial durchströmt, was den Druckverlust des Reaktors weiter verringert.
Das Reaktionsprodukt wird für eine maximale Ausbeute laufend entfernt. Dazu wird das Gasgemisch von 450 °C in einem Wärmetauscher mittels Wasser, frisch zugeführten Gasen und andere Prozessströme abgekühlt. Dabei kondensiert auch das Ammoniak und wird in einem Druckabscheider abgetrennt. Die noch nicht umgesetzten Reaktanten Stickstoff und Wasserstoff werden mittels eines Kreislaufgasverdichters wieder auf Reaktionsdruck verdichtet, mit Frischgas ergänzt und dem Reaktor zugeführt. In einer nachfolgenden Destillation wird der Ammoniak noch gereinigt.
Produkte

Der Großteil des jährlich benötigten Ammoniaks wird mit dem Haber-Bosch-Verfahren erzeugt. Die Jahresproduktion betrug 2017 etwa 150 Millionen Tonnen mit China, Indien und Russland als größten Produzenten. Aufgrund des hohen Energiebedarfs bei der Herstellung des benötigten reinen Wasserstoffs entfallen etwa 1,4 Prozent des Weltenergiebedarfs auf das Haber-Bosch-Verfahren. Die dabei erzeugten Kohlenstoffdioxidemissionen betragen etwa drei bis fünf Prozent des globalen Ausstoßes, wobei ein Teil zur Erzeugung von Harnstoff genutzt wird. Heutzutage haben, zumindest bei der Bevölkerung der Industrienationen, etwa 40 Prozent des im menschlichen Körper enthaltenen Stickstoffs schon einmal an der Haber-Bosch-Synthese teilgenommen.
Das Primärprodukt Ammoniak wird zu etwa 80 Prozent zu Dünger weiterverarbeitet, 20 Prozent entfallen auf andere Produkte. Die wichtigsten auf Ammoniak basierenden Stickstoffdünger sind neben den gasförmigen und wässrigen Lösungen von Ammoniak das Ammoniumnitrat und Harnstoff.
Die Produktion von Harnstoff in einem Hochdruckverfahren geht auf Carl Bosch und Wilhelm Meiser zurück und wurde 1922 von der BASF erstmals in Betrieb genommen. Im Jahr 2010 betrug das Produktionsvolumen 130 Millionen Tonnen. Die gesamte Weltproduktion von Salpetersäure erfolgt durch katalytische Verbrennung nach dem Ostwaldverfahren. Das Verfahren geht auf einen Vorlesungsversuch zurück, bei dem ein glühender Platindraht in ein Ammoniak-Luft-Gemisch getaucht wird, um nitrose Gase zu erzeugen. Die Weltjahresproduktion betrug 80 Millionen Tonnen im Jahr 2009. Das meistproduzierte Folgeprodukt der Salpetersäure ist Ammoniumnitrat; die Jahresproduktion betrug 2002 etwa 39 Millionen Tonnen, von denen etwa 80 Prozent zu Düngemitteln und 20 Prozent zu Sprengstoffen verarbeitet werden. Weitere Folgeprodukte wie Kaliumnitrat, partiell oder vollständig mit Ammoniak neutralisierte Phosphate wie Mono-, Di- und Ammoniumpolyphosphate, Ammoniumsulfat sowie Ammoniumnitrat-Harnstoff-Lösung sind häufig eingesetzte Dünger.
Etwa fünf Prozent der Ammoniakproduktion werden zur Herstellung von Sprengstoffen verwendet. Die in vielen Sprengstoffen vorkommenden Nitro- und Nitratgruppen basieren letztlich auf Ammoniak, das nach dem Haber-Bosch-Verfahren gewonnen wurde, darunter sind wichtige Sprengstoffe wie Trinitrotoluol und Nitroglycerin. Ungefähr zehn Prozent der Ammoniakproduktion wird für die Herstellung stickstoffhaltiger Verbindungen wie Nitrilen, Aminen und Amiden verwendet. Die Palette der Folgeprodukte ist äußerst vielfältig und reicht von Harnstoffharzen, Sulfonamiden über Nitrobenzol und dessen Folgeprodukt Anilin in die Polyurethan- und Farbstoffchemie, Caprolactam für die Produktion von Polymeren und bis hin zu Raketentreibstoffen wie Hydrazin.
Mechanismus
Elementarschritte

Der Mechanismus der Ammoniaksynthese unterteilt sich in die folgenden sieben Schritte:
- Transport der Edukte aus der Gasphase durch die Grenzschicht an die Oberfläche des Kontakts
- Porendiffusion zum Reaktionszentrum
- Adsorption der Reaktanten
- Reaktion
- Desorption der Produkte
- Rücktransport der Produkte durch das Porensystem an die Oberfläche
- Rücktransport ins Gasvolumen.
Wegen der Schalenstruktur des Katalysators sind die beiden ersten und letzten Schritte schnell gegenüber der Adsorption, der Reaktion und der Desorption. Austauschreaktionen zwischen Wasserstoff und Deuterium an Haber-Bosch-Katalysatoren finden in messbarer Geschwindigkeit noch bei Temperaturen von −196 °C statt; auch der Austausch zwischen Deuterium und Wasserstoff am Ammoniakmolekül findet bereits bei Raumtemperatur statt. Da beide Schritte schnell verlaufen, können diese nicht geschwindigkeitsbestimmend für die Ammoniaksynthese sein. Aus verschiedenen Untersuchungen ist bekannt, dass der geschwindigkeitsbestimmende Schritt der Ammoniaksynthese die Dissoziation des Stickstoffs ist.
Die Adsorption des Stickstoffs an der Katalysatoroberfläche hängt neben den Reaktionsbedingungen von der mikroskopischen Struktur der Katalysatoroberfläche ab. Eisen weist verschiedene Kristallflächen auf, deren Reaktivität höchst unterschiedlich ist. Die Fe(111)- und Fe(211)-Flächen weisen die mit Abstand höchste Aktivität auf. Die Erklärung dafür ist, dass nur diese Flächen so genannte C7-Plätze aufweisen – das sind Eisenatome mit sieben nächsten Nachbarn.
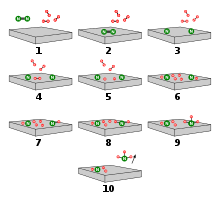
Die dissoziative Adsorption des Stickstoffs auf der Oberfläche folgt folgendem Schema, wobei S* ein Eisenatom an der Oberfläche des Katalysators bedeutet:
- N2 → S*–N2 (γ-Spezies) → S*–N2–S* (α-Spezies) → 2 S*–N (β-Spezies, Oberflächennitrid)
Die Adsorption von Stickstoff ähnelt der Chemisorption des Kohlenstoffmonoxids. Auf einer Fe(111)-Fläche führt die Adsorption von Stickstoff zunächst zu einer adsorbierten γ-Spezies mit einer Adsorptions-Energie von 24 kJmol−1 und einer N-N-Streckschwingung von 2100 cm−1. Da der Stickstoff zu Kohlenstoffmonoxid isoelektronisch ist, adsorbiert er in einer On-end-Konfiguration, in der das Molekül über ein Stickstoffatom senkrecht zur Metalloberfläche gebunden ist. Dies wurde durch Photoelektronenspektroskopie bestätigt.
Ab-initio-MO-Rechnungen haben gezeigt, dass neben der σ-Hinbindung des freien Elektronenpaars des Stickstoffs zum Metall eine π-Rückbindung aus den d-Orbitalen des Metalls in die π*-Orbitale des Stickstoffs vorliegt, welche die Eisen-Stickstoff-Bindung stärkt. Der Stickstoff im α-Zustand ist mit 31 kJmol−1 stärker gebunden. Die dadurch resultierende N-N-Bindungsschwächung konnte durch eine Verringerung der Wellenzahlen der N-N-Streckschwingung auf 1490 cm−1 experimentell belegt werden.
Ein weiteres Aufwärmen der Fe(111)-Fläche, die von α-N2 bedeckt ist, führt sowohl zu Desorption als auch zum Auftauchen einer neuen Bande bei 450 cm−1. Diese stellt eine Metall-N-Schwingung dar, den β-Zustand. Ein Vergleich mit Schwingungsspektren von Komplexverbindungen lässt den Schluss zu, dass das N2-Molekül „side-on“ gebunden ist, mit einem N-Atom in Kontakt zu einem C7-Platz. Diese Struktur wird als „Oberflächennitrid“ bezeichnet. Das Oberflächennitrid ist sehr stark an die Oberfläche gebunden. Daran addieren sich schnell Wasserstoffatome (Hads), die auf der Katalysatoroberfläche sehr beweglich sind.
Es bilden sich infrarotspektroskopisch nachgewiesene Oberflächenimide (NHad), Oberflächenamide (NH2,ad) und Oberflächen-Ammoniakate (NH3,ad), Letztere zerfallen unter NH3-Abgabe (Desorption). Die einzelnen Moleküle wurden mit Röntgenphotoelektronenspektroskopie (XPS), Hochauflösender Elektronenenergieverlustspektroskopie (HREELS) und IR-Spektroskopie identifiziert beziehungsweise zugeordnet.
-
 Reaktionsschema
Reaktionsschema
Auf Basis dieser experimentellen Befunde kann ein Reaktionsschema erstellt werden, das aus den folgenden Einzelschritten besteht:
- H2 + S* ⇌ 2 Had
- N2 + S* ⇌N2,ad
- N2,ad⇌ 2 Nad
- Nad + Had ⇌ NHad
- NHad + Had ⇌ NH2,ad
- NH2,ad + Had ⇌ NH3,ad
- NH3,ad ⇌ NH3 + S*
So wie bei jedem Haber-Bosch-Katalysator ist bei Ruthenium-Aktivkohle-Katalysatoren der geschwindigkeitsbestimmende Schritt die Stickstoff-Dissoziation. Das aktive Zentrum ist hierfür bei Ruthenium ein sogenannter B5-Platz, einer 5-fach koordinierten Position an der Ru(0001)-Oberfläche, an der zwei Ruthenium-Atome eine Stufenkante mit drei Ruthenium-Atomen der Ru(0001)-Oberfläche bilden. Die Zahl an B5-Stelle ist abhängig von Größe und Form der Ruthenium-Partikel, dem Ruthenium-Präkursor und der verwendeten Menge an Ruthenium. Die verstärkende Wirkung des basischen Trägers hat die gleiche Wirkung wie der Promotoreffekt von Alkalimetallen, der hier ebenso wie beim Eisenkatalysator zum Tragen kommt.
Energiediagramm
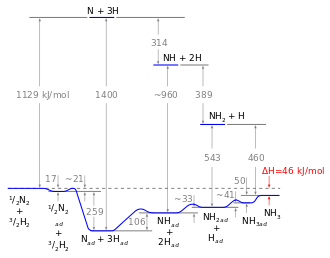
Mit dem Wissen um die Reaktionsenthalpie der einzelnen Schritte kann ein Energiediagramm erstellt werden. Mithilfe des Energiediagramms lassen sich homogene und heterogene Reaktion vergleichen: Aufgrund der hohen Aktivierungsenergie der Dissoziation von Stickstoff ist die homogene Gasphasenreaktion nicht durchführbar. Der Katalysator umgeht dieses Problem, da der Energiegewinn, der aus der Bindung von Stickstoffatomen an die Katalysatoroberfläche resultiert, die notwendige Dissoziationsenergie überkompensiert, sodass die Reaktion schlussendlich exotherm ist. Trotzdem bleibt die dissoziative Adsorption von Stickstoff der geschwindigkeitsbestimmende Schritt: nicht wegen der Aktivierungsenergie, sondern vor allem aufgrund des ungünstigen präexponentiellen Faktors der Geschwindigkeitskonstante. Die Hydrierung ist zwar endotherm, diese Energie kann jedoch leicht von der Reaktionstemperatur (etwa 700 K) aufgebracht werden.
Verfahrensvarianten
Seit der Einführung des Haber-Bosch-Verfahrens ist die Synthese von Ammoniak aus Luftstickstoff einer der weltweit wichtigsten chemischen Herstellungsprozesse geworden. Die Entwicklung von Verfahrensvarianten zu Beginn des 20. Jahrhunderts diente oft der Umgehung von Patentansprüchen der BASF. Da das Verfahren einen signifikanten Energieverbrauch erfordert, konzentrierten sich spätere Entwicklungen auf die Energieeffizienz. So betrug der durchschnittliche Energieverbrauch pro Tonne Ammoniak im Jahr 2000 noch etwa 37,4 GJ, während das thermodynamisch bedingte Minimum bei 22,4 Gigajoule pro Tonne liegt.
Casale-Verfahren
Das Casale-Verfahren wurde zu Beginn der 1920er Jahre von Luigi Casale entwickelt. Das Verfahren verwendet einen Eisenkatalysator, arbeitet aber gegenüber dem Haber-Bosch-Verfahren mit einem Druck von etwa 800 bis 1000 bar. Der Reaktor war dadurch kleiner und erlaubte durch einen internen, zentralen Wärmetauscher und die axiale Eindüsung von kaltem Gas eine gute Temperaturkontrolle.
Der hohe Betriebsdruck erlaubte die direkte Kondensation von Ammoniak ohne Absorption in Wasser. Bis 1923 errichtete Casale in Europa und den Vereinigten Staaten 15 Anlagen mit einer Kapazität von etwa 80.000 Tonnen Ammoniak pro Jahr, 1927 betrug die installierte Kapazität bereits 320.000 Tonnen pro Jahr. Zu dieser Zeit war Casale der einzige Wettbewerber der BASF. Insgesamt wurden mehr als 200 Ammoniakanlagen auf der Basis der ersten Technologie-Generation von Casale weltweit errichtet.
Uhde dual-pressure process
Anfang 2000 entwickelte Uhde einen neuen Prozess, der Anlagenkapazitäten von 3300 Tagestonnen und mehr ermöglicht. Die entscheidende Innovation des Zweidruckverfahrens ist ein nur einmal durchströmter Syntheseloop bei mittlerem Druck in Reihe mit einem konventionellen Hochdrucksynthesekreislauf. Die erste Anlage dieser Art wurde 2004 in Al-Jubail, Saudi-Arabien, erfolgreich in Betrieb genommen. Weitere Anlagen dieses Typs befinden sich in Ma’aden, Saudi-Arabien, und CFI Donaldsonville, USA.
Fauser-Verfahren
Das Fauser-Verfahren, benannt nach dem italienischen Elektroingenieur Giacomo Fauser, entsprach weitgehend dem Haber-Bosch-Verfahren, nutzte jedoch als Wasserstoffquelle die Elektrolyse von Wasser. Die Fauser-Zelle nutzte 27%ige Kalilauge als Elektrolyt und von Asbest umschlossene Anoden und Kathoden, die eine gute Trennung der entstehenden Gase sicherstellte. Das Verfahren wurde Anfang der 1920er Jahre von Montecatini eingeführt.
Mont-Cenis-Verfahren
Das Mont-Cenis-Verfahren wurde von Friedrich Uhde entwickelt und 1926 erstmals auf der Zeche Mont Cenis in Betrieb genommen. Das Verfahren, auch Niederdruckverfahren genannt, arbeitet bei Drücken von 80 bis 90 bar und einer Temperatur von 430 °C. Der verwendete Katalysator war ein Eisencyanid-Aluminiumoxid-Katalysator, der aktiver als der von Mittasch entwickelte Katalysator war. Die milderen Prozessbedingungen erlaubten den Einsatz preiswerterer Stähle für die Konstruktion der Reaktoren.
AMV-Verfahren
Imperial Chemical Industries entwickelte 1982 das AMV-Verfahren mit einem hochaktiven Eisen-Cobalt-Katalysator, der bei einem Reaktionsdruck von 100 bar und einer Temperatur von 380 °C arbeitet. Cobalt ist selbst kaum katalytisch aktiv, sondern dient der Stabilisierung des Kontakts durch Ausbildung von Spinell-Phasen mit dem Aluminiumoxid. Außerdem entstehen bei der Reduktion des Kontakts kleinere Eisenkristallite höherer Aktivität.
Eine Weiterentwicklung des Verfahrens ist das 1988 von ICI entwickelte LCA-Verfahren (Leading Concept Ammonia), das für geringere Durchsätze bei gleichem Energieeinsatz konzipiert ist. Das Kohlenstoffdioxid, das in einer einstufigen Wassergas-Shift-Reaktion anfällt, wird durch Druckwechsel-Adsorption entfernt.
Kellogg-Advanced-Ammonia-Prozess
1992 entwickelte M. W. Kellog einen Ruthenium-auf-Aktivkohle-Katalysator, der bei niedrigeren Drücken und Temperaturen arbeitet, unter dem Namen Kellogg-Advanced-Ammonia-Prozess (KAAP). Der benötigte Druck beträgt durch den aktiveren, aber teueren Rutheniumkatalysator nur noch etwa 40 bar. Als Promotoren werden Alkali- oder Erdalkalimetalle wie Caesium und Barium verwendet. Der Katalysator soll etwa 10- bis 20-mal so aktiv sein wie der herkömmliche Eisenkatalysator.
Solid-State Ammonia Synthesis
Bei der Solid-State Ammonia Synthesis (SSAS, Festkörper-Ammoniaksynthese), wird durch direkte elektrolytische Synthese von Ammoniak aus Wasser und Stickstoff unter Einsatz von elektrischer Energie der Umweg über Wasserstofferzeugung aus Wasser umgangen. Der Wirkungsgrad ist dadurch theoretisch deutlich höher, in der Laborpraxis jedoch noch weitgehend unbefriedigend. Die Bildung von Ammoniak erfolgt elektrochemisch nach folgender Gleichung:

Die Bruttogleichung der Reaktion ist:

Das Verfahren ist noch in der Entwicklung. Es gibt derzeit (Stand 2021) noch keine Produktionsanlage.
Literatur
- Hans-Erhard Lessing: Brot für die Welt, Tod dem Feind. in: Stephan Leibfried et al.(Hg): Berlins Wilde Energien – Porträts aus der Geschichte der Leibnizschen Wissenschaftsakademie. de Gruyter, Berlin, 2015, ISBN 978-3-11-037598-5
- Sandro Fehr: Die „Stickstofffrage“ in der deutschen Kriegswirtschaft des Ersten Weltkriegs und die Rolle der neutralen Schweiz. Nordhausen 2009, DNB 993295185.
- Alwin Mittasch: Geschichte der Ammoniaksynthese. Verlag Chemie, Weinheim 1951, DNB .


© biancahoegel.de
Datum der letzten Änderung: Jena, den: 29.06. 2026