Enthalpie
| Physikalische Größe | |||||||
|---|---|---|---|---|---|---|---|
| Name | Enthalpie | ||||||
| Formelzeichen | 
| ||||||
| |||||||


Die Enthalpie
(altgr.
ἐν en ‚in‘ und θάλπειν thálpein ‚erwärmen‘),
früher auch Wärmeinhalt, eines thermodynamischen
Systems
ist die Summe aus der inneren
Energie  des Systems und dem Produkt aus Druck
des Systems und dem Produkt aus Druck
 und Volumen
und Volumen
 des Systems:
des Systems:
 .
.
Sie hat die Dimension der Energie und wird in der Einheit Joule gemessen.
Die Enthalpie ist eine extensive Größe: Die Enthalpie eines Gesamtsystems ist die Summe der Enthalpien der Teilsysteme.
Die molare Enthalpie (Einheit: J/mol)
ist die auf die Stoffmenge
 bezogene Enthalpie:
bezogene Enthalpie:
 .
.
Die spezifische Enthalpie (Einheit: J/kg) ist die auf die Masse  bezogene Enthalpie:
bezogene Enthalpie:
 .
.
Die molare und die spezifische Enthalpie sind intensive Größen: Haben zwei identische Teilsysteme die gleiche molare oder spezifische Enthalpie, dann hat auch das aus ihnen gebildete Gesamtsystem diese molare bzw. spezifische Enthalpie.
Die Enthalpie
ist ebenso wie ,
und
eine Zustandsgröße;
sie wird vom aktuellen Zustand
des Systems eindeutig bestimmt und ist unabhängig von der Vorgeschichte des
Systems.
Die praktische Nützlichkeit der Enthalpie beruht darauf, dass die durch einen Prozess bewirkte Veränderung der Enthalpie eines Systems
durch den einfacheren Ausdruck

beschrieben wird, wenn der Prozess bei konstantem Druck (isobar,
 )
abläuft. Dieser Ausdruck lässt sich aber als die „Bruttoenergie“ interpretieren,
die dem System zugeführt werden muss, wenn dessen innere Energie um den Betrag
)
abläuft. Dieser Ausdruck lässt sich aber als die „Bruttoenergie“ interpretieren,
die dem System zugeführt werden muss, wenn dessen innere Energie um den Betrag
 erhöht werden soll und das System einen Teil der zugeführten Energie für die
während des Prozesses zu leistende Volumenänderungsarbeit
erhöht werden soll und das System einen Teil der zugeführten Energie für die
während des Prozesses zu leistende Volumenänderungsarbeit  verbraucht. Im Falle eines isobaren Prozesses kann man daher die aufzuwendende
„Bruttoenergie“
verbraucht. Im Falle eines isobaren Prozesses kann man daher die aufzuwendende
„Bruttoenergie“  mit der zugeführten Enthalpie
mit der zugeführten Enthalpie  identifizieren und die Vorteile nutzen, die das Rechnen mit Zustandsgrößen
bietet. Falls das System neben der Volumenänderungsarbeit keine andere Form von
Arbeit leistet, ist der Enthalpieumsatz des Prozesses gleich dem Wärmeumsatz.
identifizieren und die Vorteile nutzen, die das Rechnen mit Zustandsgrößen
bietet. Falls das System neben der Volumenänderungsarbeit keine andere Form von
Arbeit leistet, ist der Enthalpieumsatz des Prozesses gleich dem Wärmeumsatz.
Zahlreiche physikalische und chemische Prozesse finden bei konstantem Druck statt. Dies ist beispielsweise oft bei Phasenübergängen oder bei chemischen Reaktionen der Fall, insbesondere (aber nicht nur) wenn sie unter Atmosphärendruck stattfinden. Die Enthalpie ist dann eine geeignete Größe zur Beschreibung des Wärmeumsatzes dieser Prozesse.
In der theoretischen Thermodynamik ist die Enthalpie eine Fundamentalfunktion,
aus ihr lässt sich die gesamte thermodynamische Information über das System
ableiten. Voraussetzung ist jedoch, dass sie als Funktion der Variablen
Entropie
 ,
Druck
und Molzahlen
,
Druck
und Molzahlen  der im System enthaltenen chemischen Komponenten gegeben ist. Dies sind die
„natürlichen Variablen“ der Enthalpie. Sie lässt sich auch als Funktion anderer
Variablen ansetzen, enthält dann aber nicht mehr die vollständige
thermodynamische Information.
der im System enthaltenen chemischen Komponenten gegeben ist. Dies sind die
„natürlichen Variablen“ der Enthalpie. Sie lässt sich auch als Funktion anderer
Variablen ansetzen, enthält dann aber nicht mehr die vollständige
thermodynamische Information.
Die Enthalpie ist eine Legendre-Transformierte
der inneren Energie. Die innere Energie ist ebenfalls eine Fundamentalfunktion,
wenn sie als Funktion ihrer natürlichen Variablen ,
,
gegeben ist. Der Übergang zu anderen Variablensätzen erfordert die Anwendung
einer Legendre-Transformation, wenn er ohne Informationsverlust geschehen soll.
Die Transformation, die aus der inneren Energie eine Fundamentalfunktion mit den
natürlichen Variablen ,
,
erzeugt, liefert den Ausdruck  ,
also die Enthalpie. Der aus der Legendre-Transformation folgende additive Term
,
also die Enthalpie. Der aus der Legendre-Transformation folgende additive Term
 kompensiert den Informationsverlust, der sonst mit dem Variablenwechsel
verbunden wäre.
kompensiert den Informationsverlust, der sonst mit dem Variablenwechsel
verbunden wäre.
Von der Enthalpie zu unterscheiden ist die freie Enthalpie oder Gibbs-Energie, die Legendre-Transformation der Enthalpie nach der Entropie.
Einführung
Die Enthalpie ist eine mathematisch definierte abstrakte Kenngröße zur Beschreibung thermodynamischer Systeme (siehe → Thermodynamische Eigenschaften der Enthalpie). Sie lässt sich nicht unmittelbar als eine anschauliche Energiegröße eines Systems interpretieren. Unter bestimmten Bedingungen jedoch treten in einem System Energiegrößen auf, die formelmäßig mit der Enthalpie oder mit Enthalpieänderungen des Systems übereinstimmen. Sie können dann mit der Enthalpie bzw. deren Änderung identifiziert werden, was – weil die Enthalpie eine extensive Zustandsgröße ist – mathematische Vorteile bietet und mit Begriffen wie „Enthalpieinhalt“ oder „Enthalpiezufuhr“ eine kompakte und anschauliche Sprechweise bei der Beschreibung des Systems und seiner Prozesse erlaubt. Die Enthalpie wird häufig zur Beschreibung isobarer Prozesse und stationär strömender Fluide benutzt.
Enthalpie in isobaren Prozessen
Eine besonders anschauliche Bedeutung hat die Enthalpie im Falle eines bei konstantem Druck (also isobar) ablaufenden Prozesses. Da zahlreiche technische, chemische und physikalische Prozesse unter Umgebungsdruck und damit isobar ablaufen, ist diese Situation häufig anzutreffen.
Will man die innere
Energie
eines Systems erhöhen, muss man Energie von außen zuführen (Erster
Hauptsatz der Thermodynamik). Hat das System keine Möglichkeit, die
zugeführte Energie oder einen Teil davon wieder in Form von Wärme oder von mechanischer
(oder chemischer, elektrischer, magnetischer …) Arbeit
abzugeben, trägt die gesamte zugeführte Energie zur Erhöhung der inneren Energie
bei. Wird die Energie beispielsweise in Form einer Wärmemenge  zugeführt, lautet die Energiebilanz für ein solches System einfach
zugeführt, lautet die Energiebilanz für ein solches System einfach  .
.
In der Regel jedoch ist die Erhöhung der inneren Energie mit einer
Volumenvergrößerung verbunden, etwa infolge thermischer
Expansion bei Temperaturerhöhung, im Zuge eines Phasenübergangs
oder bei einer chemischen
Reaktion mit Gasentwicklung. Wird die Volumenzunahme nicht verhindert (das
wäre zum Beispiel bei starr eingespannten Systemen oder bei chemischen
Reaktionen in einem starren Behälter der Fall), dehnt sich das System um das
Volumen  gegen den auflastenden Umgebungsdruck
aus und leistet dabei die Volumenänderungsarbeit
gegen den auflastenden Umgebungsdruck
aus und leistet dabei die Volumenänderungsarbeit
 ,
die nun nicht mehr zur Erhöhung der inneren Energie zur Verfügung steht:
,
die nun nicht mehr zur Erhöhung der inneren Energie zur Verfügung steht:

oder umgestellt

Die Änderung der Enthalpie des Systems ist andererseits, gemäß deren Definition und unter Anwendung der Produktregel
 ,
,
was sich im Spezialfall konstanten Drucks ()
reduziert auf
 .
.
Vergleich der markierten Ausdrücke liefert
 .
.
Geht also ein System bei konstantem Druck von einem Anfangs- in einen Endzustand über und tritt dabei keine andere Form von Arbeit auf als Volumenänderungsarbeit, dann ist die Änderung der Enthalpie des Systems zahlenmäßig gleich der dem System zugeführten Wärmemenge.
In der älteren Literatur wurde die Enthalpie deshalb auch als „Wärmeinhalt“ des Systems bezeichnet. In heutiger Sprechweise ist das nicht mehr üblich, weil man dem resultierenden Energieinhalt des Systems nicht ansehen kann, ob die Energie als Wärme oder als Arbeit zugeführt wurde. Näheres hierzu folgt im nächsten Abschnitt.
Die genannte Einschränkung auf Systeme, die keine andere Arbeit leisten als Volumenänderungsarbeit, ist ebenso wichtig wie die Beschränkung auf konstanten Druck. Ein häufig anzutreffendes Beispiel für Systeme, die auch eine andere Form von Arbeit leisten können, sind galvanische Zellen. Diese können elektrische Arbeit leisten, und die von ihnen umgesetzte Wärme ist nicht identisch mit der Änderung ihrer Enthalpie.
Die Verwendung der Enthalpie ist nicht auf isobare Prozesse beschränkt. Im
Falle nicht-isobarer Prozesse ist die Enthalpieänderung bei Wärmezufuhr jedoch
komplizierter:  .
.
Enthalpie als Zustandsgröße
Zustands- und Prozessgrößen
Eine Zustandsgröße ist durch den aktuellen Zustand des Systems eindeutig festgelegt. Sie ist insbesondere unabhängig von der Vorgeschichte des Systems, also von dem Prozess, über den es in den vorliegenden Zustand gelangte. Beispiele sind Temperatur, Druck, innere Energie und Enthalpie.
Eine Prozessgröße beschreibt den Vorgang, der das System von einem Zustand in einen anderen überführt. Gibt es verschiedene Prozessführungen, die von einem gegebenen Anfangszustand zu einem gegebenen Endzustand führen, können die jeweiligen Prozessgrößen trotz fixierter Anfangs- und Endzustände verschieden sein. Beispiele sind der Wärmestrom, den das System während des Prozesses mit seiner Umgebung austauscht, oder die mechanische Arbeit, die es mit der Umgebung austauscht.
Wärmeinhalt und Enthalpieinhalt
Wird einem System ein Wärmestrom zugeführt, ist man intuitiv versucht, sich dabei eine „Wärmemenge“ vorzustellen, die in das System fließt, und deren angesammelte Gesamtmenge eine vermeintliche „Zustandsgröße Wärmeinhalt“ des Systems darstellt. Diese Vorstellung ist jedoch nicht haltbar. Dem System kann auch mechanische Arbeit zugeführt werden, deren Summe dann konsequenterweise als „Arbeitsinhalt“ angesehen werden müsste. Da sich der im System befindlichen Energie aber nicht mehr ansehen lässt, ob sie als Wärme oder Arbeit zugeführt wurde, ist es nicht sinnvoll, sie begrifflich in einen „Wärmeinhalt“ und einen „Arbeitsinhalt“ aufzuteilen. Außerdem gibt es in der Regel verschiedene Wege, auf denen der neue Zustand erreicht werden kann, und die dem System insgesamt zugeführte Energie kann je nach Prozessführung unterschiedlich auf Wärme- und Arbeitszufuhr aufgeteilt sein. Das System könnte dann im neuen Zustand je nach durchlaufenem Prozessweg unterschiedliche „Wärmeinhalte“ und „Arbeitsinhalte“ aufweisen, so dass diese auch keine Zustandsgrößen sein könnten. Unter „Wärme“ versteht man deshalb im Sprachgebrauch der Thermodynamik ausschließlich die Prozessgröße „Wärmestrom“, der Begriff „Wärmeinhalt“ wird nicht mehr benutzt.
Die innere Energie und die Enthalpie sind im Gegensatz zum „Wärmeinhalt“
Zustandsgrößen, und da sie außerdem extensive (d.h. mengenartige) Größen
sind, kann anschaulich vom Inhalt an innerer Energie sowie vom Enthalpieinhalt
des Systems gesprochen werden. Beide ändern sich bei Zufuhr einer Wärmemenge,
und zwar erhöht sich bei isochorer
Prozessführung (also konstant gehaltenem Volumen) der Inhalt an innerer
Energie genau um die zugeführte Wärmemenge, ,
und bei isobarer Prozessführung (konstant gehaltenem Druck) ändert sich
der Enthalpieinhalt genau um die zugeführte Wärmemenge, .
Diese beiden Zustandsgrößen
beziehungsweise
leisten für die jeweilige Prozessart also genau das, wofür man intuitiv den
Begriff des „Wärmeinhalts“ benutzt hätte, und ersetzen diesen.
Die Zustandsgrößen haben außerdem den Vorteil, dass der Unterschied an innerer Energie oder Enthalpie zwischen zwei Zuständen eines Systems allein aus der Kenntnis der beiden Zustände bestimmt werden kann und nicht von der Art abhängt, wie die Zustandsänderung erfolgte. Die Bestimmung von Prozessgrößen, beispielsweise wie sich die während der Zustandsänderung ausgetauschte Energie in Wärme und Arbeit aufteilt, erfordert in der Regel zusätzliche Kenntnis über Details des Änderungsprozesses.
Enthalpieinhalt und Enthalpiezufuhr
Wird ein System durch einen geeigneten (isochoren) Prozess von einem Zustand
 in einen Zustand
in einen Zustand  überführt, so hat es im Anfangszustand die Enthalpie
überführt, so hat es im Anfangszustand die Enthalpie  und im Endzustand die Enthalpie
und im Endzustand die Enthalpie  .
Was bedeutet der Enthalpie-Unterschied, ausgedrückt durch messbare physikalische
Größen?
.
Was bedeutet der Enthalpie-Unterschied, ausgedrückt durch messbare physikalische
Größen?
Für einen infinitesimal kleinen Enthalpieunterschied gilt (s.o.)
- ,
so dass bei gegebenem aktuellen Volumen
eine geeignete Kombination von Wärmezufuhr
und Druckänderung  den Enthalpieinhalt
des Systems um den Betrag
ändert. Es ist naheliegend,
als Enthalpiezufuhr und die zeitliche Zufuhr-Rate
den Enthalpieinhalt
des Systems um den Betrag
ändert. Es ist naheliegend,
als Enthalpiezufuhr und die zeitliche Zufuhr-Rate  als Enthalpiestrom zu bezeichnen. Ein endlich großer Enthalpieunterschied
als Enthalpiestrom zu bezeichnen. Ein endlich großer Enthalpieunterschied  ist das Integral über .
ist das Integral über .
Ist die Enthalpie
unbekannt und soll sie messtechnisch bestimmt werden, können die Größen ,
und
während des Prozesses gemessen und von
ausgehend aufsummiert werden.
Im isobaren Fall ist
und es genügt, die Wärmezufuhr
zu messen und aufzusummieren, beispielsweise in einem geeigneten Kalorimeter. Der
Zusammenhang zwischen der isobaren Enthalpiezufuhr und der damit einhergehenden
Temperaturänderung wird durch die Wärmekapazität bei konstantem Druck  beschrieben (siehe Abschnitt Enthalpie
und Wärmekapazität). Ist
für ein isobares System bekannt, ist damit auch die einer gegebenen
Temperaturdifferenz entsprechende Enthalpiedifferenz bekannt (siehe Beispiel
2).
beschrieben (siehe Abschnitt Enthalpie
und Wärmekapazität). Ist
für ein isobares System bekannt, ist damit auch die einer gegebenen
Temperaturdifferenz entsprechende Enthalpiedifferenz bekannt (siehe Beispiel
2).
Sind umgekehrt
und
bekannt, gibt die Enthalpiedifferenz Hinweise auf mögliche Prozessführungen und
beschreibt im isobaren Fall, welcher Wärmeumsatz zu erwarten ist. Für zahlreiche
Systeme können die zu den verschiedenen Zuständen gehörigen Enthalpien
einschlägigen Tabellenwerken entnommen werden.
Ist die Enthalpie des Endzustands kleiner als die des Anfangszustands, muss
während des Prozesses die der Differenz entsprechende Enthalpie  abgeführt werden – der Prozess ist exotherm.
Im Falle eines isobaren Prozesses ist die abzuführende Wärmemenge
abgeführt werden – der Prozess ist exotherm.
Im Falle eines isobaren Prozesses ist die abzuführende Wärmemenge  zahlenmäßig gleich der ermittelten Enthalpiedifferenz.
zahlenmäßig gleich der ermittelten Enthalpiedifferenz.
Ist die Enthalpie des Endzustands größer als die des Anfangszustands, muss die der Differenz entsprechende Enthalpie zugeführt werden – der Prozess ist endotherm. Im Falle eines isobaren Prozesses ist die zuzuführende Wärmemenge zahlenmäßig gleich der ermittelten Enthalpiedifferenz.
Da Anfangs- und Endenthalpie nur vom Anfangs- und Endzustand abhängen, nicht aber vom dazwischenliegenden Prozess, kann bei der rechnerischen Ermittlung einer unbekannten Enthalpiedifferenz der reale Prozess durch einen leichter behandelbaren Prozess oder sogar durch eine über Hilfszustände mit bekannten Enthalpien führende Prozesskette ersetzt werden, solange diese dieselben beiden Zustände miteinander verbindet. Praktische Beispiele folgen weiter unten.
In den meisten Fällen ist der absolute Enthalpieinhalt eines Systems nicht von Belang und nur der vom Prozess bewirkte Enthalpieunterschied von Interesse. In diesem Fall hat man die Freiheit, den Nullpunkt für die Messung der Enthalpie beliebig zu wählen.
Man beachte, dass von der zugeführten Energie ein Teil als Verschiebungsarbeit in die Umgebung abgeführt wird und nur der Rest im System selbst verbleibt und dessen Energieinhalt vermehrt. Die zugeführte Enthalpie hingegen denkt man sich in vollem Umfang im System gespeichert. Diese Sprechweise ist zulässig, weil die Enthalpie eine Zustandsgröße des Systems ist. Lässt man den Prozess umgekehrt ablaufen, erhält man sowohl die gesamte dem System zugeführte Energie als auch die gesamte dem System zugeführte Enthalpie wieder zurück. Das System gewinnt dabei die in die Umgebung abgegebene Energie als negative Verschiebungsarbeit wieder zurück, die Enthalpie denkt man sich aus dem Enthalpieinhalt des Systems stammend. Es zeigt sich erneut, dass die Enthalpie nicht eine bestimmte konkrete „Art von Energie“ ist, sondern eine abstrakte Größe, die jedoch eine sehr nützliche Sprechweise erlaubt.
Für die Enthalpie gilt im Allgemeinen kein Erhaltungssatz. Man betrachte als Beispiel einen thermisch isolierten Behälter konstanten Volumens, in dem eine chemische Reaktion abläuft. Die Änderung der Enthalpie

reduziert sich wegen  (kein Energieaustausch mit der Umgebung; innere Energie unterliegt der
Energieerhaltung) und
(kein Energieaustausch mit der Umgebung; innere Energie unterliegt der
Energieerhaltung) und  (Volumenkonstanz des Behälters) auf
(Volumenkonstanz des Behälters) auf
 .
.
Obwohl das System innerhalb des Behälters weder Energie noch Materie mit der Umgebung austauscht, ändert sich seine Enthalpie, wenn sich der Druck im Behälter im Zuge der chemischen Reaktion ändert. Der Begriff „Enthalpiezufuhr“ wäre in diesem Fall, wenn man ihn überhaupt verwenden wollte, nur als eine Sprechweise für „Enthalpieerhöhung“ zu verstehen. Es wird jedoch keine Enthalpie aus der Umgebung abgezogen und in den Behälter transportiert.
Im isobaren Fall, in dem die Enthalpieänderung zahlenmäßig identisch mit der umgesetzten Wärmemenge ist, gilt für die Enthalpie der Energieerhaltungssatz, dem die Wärmeenergie unterliegt.
Beispiele
Beispiel 1: Enthalpieänderung bei einem Phasenübergang
Gegeben sei flüssiges Wasser im Gleichgewicht mit seinem Dampf. Die Temperatur beider Phasen betrage 10 °C, der Druck in beiden Phasen sei der Sättigungsdampfdruck bei der gegebenen Temperatur (ca. 1228 Pa). Einer Wasserdampftafel lässt sich entnehmen, dass bei dieser Temperatur und diesem Druck
- die spezifische Enthalpie des flüssigen Wassers
 und
und - die spezifische Enthalpie des Wasserdampfes
 beträgt.
beträgt.
Um Wasser bei der gegebenen Temperatur und konstantem Druck zu verdampfen,
muss die spezifische Enthalpie von  auf
auf  erhöht werden, es müssen also
erhöht werden, es müssen also

an spezifischer Enthalpie zugeführt werden. Da es sich um einen isobaren
Vorgang handelt, kann die Enthalpieerhöhung durch Zuführung der spezifischen
Wärmemenge  erzielt werden. Dieser Energiebetrag ist die so genannte spezifische
Verdampfungswärme oder spezifische Verdampfungsenthalpie des
Wassers bei 10 °C.
erzielt werden. Dieser Energiebetrag ist die so genannte spezifische
Verdampfungswärme oder spezifische Verdampfungsenthalpie des
Wassers bei 10 °C.
Man beachte auch die hierbei verwendete Sprechweise: Die Enthalpie ist eigentlich als Zustandsgröße definiert und stellt damit eine Eigenschaft des Systems dar. Die im Beispiel ermittelten 2477 kJ/kg sind eine Enthalpiedifferenz zwischen zwei Systemzuständen. Man nennt sie aber ebenfalls eine Enthalpie, worin sich wieder die Vorstellung spiegelt, man habe es mit einer mengenartigen Größe zu tun und müsse eine gewisse „Menge an Enthalpie“ zuführen, um den „Enthalpieinhalt“ des Systems um die betreffende Menge zu erhöhen.
Der Wasserdampftafel lässt sich außerdem entnehmen, dass bei dieser Temperatur und diesem Druck
- das spezifische
Volumen des flüssigen Wassers
 und
und - das spezifische Volumen des Wasserdampfs
 beträgt.
beträgt.
Die Zunahme des spezifischen Volumens ist also

und die durch diese Volumenzunahme geleistete spezifische Volumenänderungsarbeit beträgt
 .
.
Von den als Wärme zugeführten
werden also  wieder an die Umgebung abgegeben und die restlichen
wieder an die Umgebung abgegeben und die restlichen  erhöhen die innere Energie des Systems.
erhöhen die innere Energie des Systems.
Beispiel 2: Vorteil der Enthalpie als Zustandsfunktion
Dieses Beispiel zeigt den Vorteil, der sich aus dem Umstand ziehen lässt, dass die Enthalpie eine Zustandsfunktion ist (im Gegensatz zum „Wärmeinhalt“). Gegeben sei eine Knallgasreaktion, bei der aus je zwei Atomen Wasserstoff und einem Atom Sauerstoff ein Wassermolekül gebildet wird:

Einem einschlägigen Tabellenwerk sei die Angabe entnommen, dass bei einer Temperatur von 25 °C und einem Druck von einer Atmosphäre die Enthalpie von einem Mol des entstandenen Wassers um 285,84 kJ geringer ist als die Enthalpie der Ausgangsstoffe auf der linken Seite:

Wie groß ist der molare Enthalpieunterschied beider Seiten, wenn der Druck
bei einer Atmosphäre bleibt, aber die Temperatur auf 100 °C erhöht wurde? Als
Zusatzinformation seien aus Experimenten die folgenden molaren Wärmekapazitäten
bei konstantem Druck
bekannt (siehe Abschnitt Enthalpie
und Wärmekapazität), jeweils als Mittelwert über den Temperaturbereich von
25 °C bis 100 °C:






Anstelle des zu untersuchenden Originalprozesses, der vom Ausgangszustand
 direkt in den Endzustand
direkt in den Endzustand  führt, wird rechnerisch eine Kette von Ersatzprozessen mit bekannten
Eigenschaften betrachtet, welche dieselben beiden Zustände miteinander
verbindet. Dies ist erlaubt, weil der Enthalpieunterschied zwischen Anfangs- und
Endzustand nur von diesen Zuständen selbst bestimmt wird und nicht von den
speziellen Prozessen, die beide ineinander überführen.
führt, wird rechnerisch eine Kette von Ersatzprozessen mit bekannten
Eigenschaften betrachtet, welche dieselben beiden Zustände miteinander
verbindet. Dies ist erlaubt, weil der Enthalpieunterschied zwischen Anfangs- und
Endzustand nur von diesen Zuständen selbst bestimmt wird und nicht von den
speziellen Prozessen, die beide ineinander überführen.
|
|

|
|

|

| |

|
| |
|
|
| |

|

| |
|
|
| |

|
| |
|
|
| |

|

|

|
In einem ersten rechnerischen Schritt wird der Wasserstoff von 100 °C auf 25 °C abgekühlt. Dabei ändert sich seine molare Enthalpie um
 .
.
Dann wird das halbe Mol Sauerstoff auf dieselbe Temperatur abgekühlt, seine Enthalpie ändert sich um
 .
.
Die molare Reaktionsenthalpie bei 25 °C ist bekannt:
 .
.
Das Erwärmen des entstandenen Wassers auf 100 °C erfordert die Zufuhr von
 .
.
Die Summe der molaren Enthalpieänderungen während des Ersatzprozesses

ist identisch mit der molaren Enthalpieänderung auf dem direkten Prozesspfad, also ist
 .
.
Stationär strömende Fluide
Über isobare Prozesse hinaus ist die Enthalpie auch eine nützliche Größe bei der Beschreibung von Systemen im Fließgleichgewicht wie zum Beispiel Wärmekraftmaschinen oder Drosseln, wo sie zur anschaulichen Beschreibung des Energieflusses dienen kann.
Man betrachte eine Wärmekraftmaschine, die von einer stationären Strömung
eines Arbeitsfluids durchflossen wird. Im
Zuleitungsrohr mit der Querschnittsfläche  habe der im zufließenden Fluid herrschende Druck
habe der im zufließenden Fluid herrschende Druck  im betrachteten Zeitraum das Volumen
im betrachteten Zeitraum das Volumen  in die Maschine gedrückt. Das nachfließende Fluid hat dabei die Kraft
in die Maschine gedrückt. Das nachfließende Fluid hat dabei die Kraft  auf das Volumen ausgeübt und es um die Strecke
auf das Volumen ausgeübt und es um die Strecke  verschoben. Die am Volumen geleistete mechanische Arbeit ist daher
verschoben. Die am Volumen geleistete mechanische Arbeit ist daher  .
Stellt man noch die innere Energie
.
Stellt man noch die innere Energie  des Testvolumens in Rechnung und vernachlässigt dessen kinetische Energie, dann
wurde der Maschine die Energie
des Testvolumens in Rechnung und vernachlässigt dessen kinetische Energie, dann
wurde der Maschine die Energie

zugeführt. Entsprechende Überlegungen gelten für ein im gleichen Zeitraum
unter dem Druck  durch den Querschnitt
durch den Querschnitt  des Abflussrohres fließendes Volumen
des Abflussrohres fließendes Volumen  .
Setzt man der Allgemeinheit halber noch eine der Maschine im betrachteten
Zeitraum zugeführte Wärmemenge
.
Setzt man der Allgemeinheit halber noch eine der Maschine im betrachteten
Zeitraum zugeführte Wärmemenge  und eine von der Maschine geleistete Nutzarbeit
und eine von der Maschine geleistete Nutzarbeit  an, so lautet die Energiebilanz einfach
an, so lautet die Energiebilanz einfach
 .
.
In einem solchen stationär strömenden Arbeitsfluid lässt sich der Term  anschaulich interpretieren als die vom Fluid kontinuierlich geleistete (und
damit sozusagen „transportierte“) Verschiebearbeit.
anschaulich interpretieren als die vom Fluid kontinuierlich geleistete (und
damit sozusagen „transportierte“) Verschiebearbeit.
Ein wichtiger Sonderfall ist eine Drossel,
also ein Rohr, in dem eine Engstelle oder ein poröser Pfropfen den Druck des
strömenden Fluids von
auf
vermindert. Ist die Rohrwand adiabatisch ausgeführt ( )
und wird dem System keine mechanische Arbeit entzogen (
)
und wird dem System keine mechanische Arbeit entzogen ( ),
dann lautet die Energiebilanz der Drossel
),
dann lautet die Energiebilanz der Drossel
 ,
,
die Drosselung ist also ein isenthalper
Vorgang. Die Größen ,
und
des Testvolumens haben in der Regel nach der Drosselung andere Werte als vor der
Drosselung, die Änderungen hängen aber in solcher Weise zusammen, dass die
Größenkombination
unverändert bleibt.
Gleichheit der Enthalpien der Testvolumina kann dabei nur für einen Fluidzustand vor der Drosselung und einen nach der Drosselung festgestellt werden. Es kann nicht davon gesprochen werden, dass die Enthalpie der Fluidvolumina entlang der gesamten Drosselstrecke konstant sei, da an der Drosselstelle in der Regel Nichtgleichgewichtszustände vorliegen, für die keine Enthalpie definiert ist. Falls die kinetischen Energien beiderseits der Drosselstelle nicht vernachlässigbar klein sind, genügt es auch, wenn ihr Unterschied vernachlässigbar klein ist, weil sie sich dann auf beiden Seiten der Energiebilanz fortkürzen.
Ist das Arbeitsfluid insbesondere ein ideales
Gas, sind ,
,
und damit auch
nur von der Temperatur abhängig. Gleichheit der Enthalpien bedeutet daher in
diesem Fall auch Gleichheit der Temperaturen vor und nach der Drosselung: Ein
ideales Gas erfährt durch die Drosselung keine Temperaturveränderung. Bei
nicht-idealen Fluiden kann sich je nach Art des Fluids und den
Prozessbedingungen eine Temperaturzunahme oder -abnahme ergeben (siehe → Joule-Thomson-Effekt).
Enthalpie in isobaren physikalischen und chemischen Prozessen
Zahlreiche Prozesse aus der Physik (z.B. Phasenübergänge) oder aus der Chemie (z.B. chemische Reaktionen) laufen unter konstantem Druck ab. Die Enthalpie erlaubt in diesen Fällen eine einfache Beschreibung und Berechnung des Wärmeumsatzes.
Standardbildungsenthalpie
Wie bereits erwähnt, kann zur Berechnung des Enthalpieunterschieds zwischen zwei Zuständen ein beliebiger Prozess verwendet werden, der die beiden Zustände miteinander verbindet. Man kann beispielsweise bei einer chemischen Reaktion die Ausgangsstoffe gedanklich in ihre Elemente zerlegen und diese wieder zu den Produktstoffen zusammensetzen. Die dabei jeweils aufzuwendende oder abzuführende Enthalpie ist die so genannte Bildungsenthalpie des betreffenden Stoffes. Die Bildungsenthalpien sind temperatur- und druckabhängig. Die Bildungsenthalpien, die unter Standardbedingungen umgesetzt werden, sind die Standardbildungsenthalpien.
Die molare Standardbildungsenthalpie (meist kurz
Standardbildungsenthalpie) ist die Enthalpie, die bei der Bildung von
einem Mol einer
Substanz aus der allotropisch
stabilsten Form der reinen Elemente unter Standardbedingungen (100 kPa und
25 °C) frei wird (negatives Vorzeichen) oder zur Bildung erforderlich ist
(positives Vorzeichen). Sie wird üblicherweise in Kilojoule pro Mol angegeben.
Die Größe wird  formuliert (
formuliert ( von engl. formation steht darin für Bildung; die hochgestellte Null für
Standardbedingungen; das Delta für die Differenz).
von engl. formation steht darin für Bildung; die hochgestellte Null für
Standardbedingungen; das Delta für die Differenz).
Ist sie negativ, handelt es sich um eine exotherme Reaktion und bei der Bildung der Substanz aus den Elementen wird Energie frei (Bildungswärme). Ist sie dagegen positiv, handelt es sich um eine endotherme Reaktion und es muss Energie zur Bildung der Substanz aus ihren Ausgangselementen aufgewendet werden. Stark negative Werte der Standardbildungsenthalpie sind ein Kennzeichen chemisch besonders stabiler Verbindungen (d.h. bei ihrer Bildung wird viel Energie frei und zur Zerstörung der Bindungen muss auch wieder viel Energie aufgewendet werden). Die Standardbildungsenthalpie der chemischen Elemente in ihrem stabilsten Zustand (H2, He, Li, …) ist per Definition auf 0 kJ/mol festgesetzt.
Sind die Standardbildungsenthalpien der an einer chemischen Reaktion beteiligten Stoffe bekannt, so lässt sich die Reaktionsenthalpie dieser Reaktion unter Standardbedingungen leicht berechnen. Sie ist die Differenz zwischen den Standardbildungsenthalpien der Reaktionsprodukte („Produkte“) einerseits und der Ausgangsstoffe (Reaktanten; „Edukte“) andererseits (Satz von Hess):

Alle Werte beziehen sich auf das thermodynamische Gleichgewicht, da sonst die Temperatur nicht definiert wäre.
Umgekehrt kann die Standardbildungsenthalpie unter Zuhilfenahme des Satzes von Hess aus Enthalpien von Reaktionen bestimmt werden, bei denen der jeweilige Stoff als Edukt oder Produkt teilnimmt. Wenn keine experimentellen Daten vorliegen, kann eine Abschätzung der Standardbildungsenthalpien auch mit Gruppenbeitragsmethoden abgeschätzt werden. Hierfür eignet sich besonders die Inkrement-Methode nach Benson.
Anorganische Stoffe
| Chemische Formel | Stoff |
(kJ/mol) |
|---|---|---|
| H2O (g) | Wasser (gasförmig) | −241,83 |
| H2O (l) | Wasser (flüssig) | −285,83 |
| CO2 (g) | Kohlendioxid | −393,50 |
| NH3 (g) | Ammoniak | −45,94 |
Organische Stoffe
| Summenformel | Stoff |
(kJ/mol) |
|---|---|---|
| CH4 (g) | Methan | −74,87 |
| C2H4 (g) | Ethylen | +52,47 |
| C2H6 (g) | Ethan | −83,8 |
Enthalpie in der Physik (Thermodynamik)
Die Thermodynamik beschreibt im engeren Sinne nur die intermolekularen Kräfte, also die energetischen Beziehungen (Phasenzustände bzw. deren Änderungen) zwischen den einzelnen Molekülen eines Stoffs.
Verdampfungsenthalpie / Kondensationsenthalpie
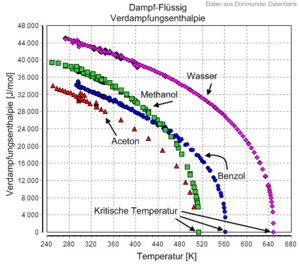
Die molare Verdampfungsenthalpie  ist die Enthalpie, die erforderlich ist, um ein Mol einer Substanz isotherm
und isobar vom flüssigen in den gasförmigen Zustand zu bringen. Die
Verdampfungsenthalpie ist vom Stoff sowie von
Temperatur und Druck abhängig, wobei sie immer positiv ist und ihr Vorzeichen
daher in der Regel nicht angegeben wird.
ist die Enthalpie, die erforderlich ist, um ein Mol einer Substanz isotherm
und isobar vom flüssigen in den gasförmigen Zustand zu bringen. Die
Verdampfungsenthalpie ist vom Stoff sowie von
Temperatur und Druck abhängig, wobei sie immer positiv ist und ihr Vorzeichen
daher in der Regel nicht angegeben wird.
Die molare Kondensationsenthalpie  ist die Enthalpie, die frei wird, wenn ein Mol einer Substanz kondensiert, wobei diese
wieder isotherm und isobar vom gasförmigen in den flüssigen Aggregatzustand
übergeht. Sie trägt immer ein negatives Vorzeichen.
ist die Enthalpie, die frei wird, wenn ein Mol einer Substanz kondensiert, wobei diese
wieder isotherm und isobar vom gasförmigen in den flüssigen Aggregatzustand
übergeht. Sie trägt immer ein negatives Vorzeichen.
Die Verdampfungsenthalpie nimmt mit steigender Temperatur ab und wird bei
Erreichen des kritischen
Punkts Null, da dort keine Unterscheidung von Flüssigkeit und Gas mehr
möglich ist. In der Regel werden in Tabellenwerken Verdampfungsenthalpie-Daten
entweder auf 25 °C bezogen oder für die verschiedenen
Temperatur-Druck-Kombinationen entlang der Siedepunktskurve
tabelliert, es gilt dabei immer  .
.
Bei Gemischen oder Lösungen von Stoffen addieren sich die Enthalpien im Verhältnis ihrer Mischungsanteile.
Sofern für eine Substanz keine Verdampfungsenthalpiewerte verfügbar sind, kann man diese für beliebige Temperaturen mit Hilfe der Clausius-Clapeyron-Gleichung berechnen, wenn die Temperaturabhängigkeit der Dampfdruckkurve bei der betrachteten Temperatur bekannt ist.
In seltenen Fällen wurden Werte für Verdampfungsenthalpien tabelliert. Die Verdampfungsenthalpie kann immer dann durch Differenzbildung aus den thermodynamischen Daten abgeleitet werden, wenn Standardbildungsenthalpie-Werte für den flüssigen und gasförmigen Aggregatzustand bekannt sind, z.B. für Wasser, Schwefelkohlenstoff, Methanol, Ethanol, Ameisensäure, Essigsäure, Brom in obenstehender Tabelle.
Beispiel Kochsalz:
- Verdampfungsenthalpie ∆VH = +170 kJ/mol (bei 1465 °C, Tabellenwert)
- Zur praktischen Berechnung der Verdampfungswärme werden folgende Tabellenwerte für die Bildungsenthalpie verwendet:
-
NaCl (schmelze) 
NaCl (g) −386 kJ/mol −182 kJ/mol Bildungsenthalpien (25 °C)  Verdampfungsenthalpie ∆VH = +204 kJ/mol
Verdampfungsenthalpie ∆VH = +204 kJ/mol
Der Unterschied der zwei Werte 170 und 204 liegt im üblichen Rahmen.
Sublimationsenthalpie
Die Sublimation beschreibt den Übergang eines Feststoffs in die Gasphase unter Umgehung der flüssigen Schmelzphase (technische Anwendung bei der Gefriertrocknung). Die Sublimationsenthalpie wird teilweise in Tabellenwerken aufgeführt. Prinzipiell dürfen hierzu bei gleicher Bezugstemperatur auch Schmelz- und Verdampfungsenthalpie zusammengefasst werden:
- Sublimationsenthalpie = Schmelzenthalpie + Verdampfungsenthalpie.
Nicht zu verwechseln ist die Sublimationsenthalpie mit der Sublimationswärme. Diese entspricht der Summe aus Schmelzwärme und Verdampfungswärme.
Die Sublimationsenthalpie kann immer dann aus den thermodynamischen Daten abgeleitet werden, wenn Standardbildungsenthalpie-Werte für den festen und gasförmigen Aggregatzustand bekannt sind.
- Sublimationsenthalpie Kochsalz: 211 kJ/mol (25 °C, Tabellenwert)
- Berechnung:
-
NaCl (s) NaCl (g) −411 kJ/mol −182 kJ/mol Bildungsenthalpien (25 °C)
Sublimationsenthalpie +229 kJ/mol
Anm.: Das Beispiel zeigt, dass man prinzipiell auch Vorgänge berechnen kann, die praktisch kaum durchführbar sind. Die „Sublimationsenthalpie von elementarem Kohlenstoff“ wurde so „ermittelt“.
| Sublimationenthalpien einiger Stoffe | ||
|---|---|---|
| Stoffzeichen | Stoffe | Sublimationsenthalpie (kJ/mol) |
| Na | Natrium | 108 |
| K | Kalium | 89 |
| Rb | Rubidium | 82 |
| Cs | Caesium | 78 |
| Mg | Magnesium | 150 |
| Ca | Calcium | 192 |
| Sr | Strontium | 164 |
| Ba | Barium | 176 |
| I2 | Iod | 62,4 |
| C10H8 | Naphthalin | 72,6 |
| CO2 | Kohlenstoffdioxid | 26,1 |
Schmelzenthalpie / Kristallisationsenthalpie
Nach dem Erwärmen einer festen Substanz bis zu ihrer Schmelzpunkttemperatur wird bei dieser Temperatur Schmelzwärme aufgenommen, ohne dass die Temperatur weiter ansteigt. Diese Form von Wärme wird latente Wärme genannt, weil diese keine Temperaturänderung bewirkt. Bei ionischen Feststoffen entstehen bei der Phasenumwandlung fest/flüssig Salzschmelzen mit leicht beweglichen Ionen (techn. Anwendung bei der Schmelzflusselektrolyse). Kochsalz schmilzt bei 800 °C.
Schmelzenthalpien sind nur selten in Tabellenwerken erfasst.
Die Schmelzenthalpie kann immer dann aus den thermodynamischen Daten abgeleitet werden, wenn Standardbildungsenthalpie-Werte für den festen und flüssigen Aggregatzustand bekannt sind.
- Schmelzenthalpie Kochsalz: 28; 30,2 kJ/mol (800 °C, Tabellenwerte)
- Zur praktischen Berechnung der Schmelzwärme werden folgende Tabellenwerte verwendet:
-
NaCl (s) NaCl (Schmelze) −411 kJ/mol −386 kJ/mol Bildungsenthalpien (25 °C) NaCl (s) Na+ (Schmelze) + Cl− (Schmelze) −411 kJ/mol ca. −215 kJ/mol ca. −170 kJ/mol Bildungsenthalpien (25 °C)
Schmelzenthalpie +25 kJ/mol (NaCl, 25 °C)
Bei der Umkehrung dieses Prozesses, der Kristallisation aus der Schmelze, können die Ionen eines Salzes sich direkt zu ihrem festen Kristallgitter zusammenschließen. Während des Ausscheidens von Kochsalzkristallen aus der Schmelze werden −25,2 kJ/mol Kristallisationsenthalpie (bzw. 29±1 kJ/mol bei 800 °C) freigesetzt.
Erfahrungsgemäß können „unterkühlte Salzschmelzen“ durch eine spontan einsetzende Kristallisation erhebliche Wärmemengen freisetzen. (Anwendung: Heizkissen).
Gitterenthalpie
Nach einer gängigen Definition ist die Gitterenergie
diejenige Energie, die im Vakuum (d.h. bei äußerem Druck  )
aufgewendet werden muss, um einen kristallinen ionischen Feststoff in die
Gasphase zu überführen (d.i. Sublimationsenergie), ihn also in die
gasförmigen Ionen zu separieren. Die Gitterenergie und die Gitterenthalpie
unterscheiden sich qualitativ. Die Gitterenergie ist eine innere Energie,
während die Gitterenthalpie eine Enthalpie ist. Die Gitterenthalpie
berücksichtigt also zusätzlich die zu leistende Volumenarbeit
)
aufgewendet werden muss, um einen kristallinen ionischen Feststoff in die
Gasphase zu überführen (d.i. Sublimationsenergie), ihn also in die
gasförmigen Ionen zu separieren. Die Gitterenergie und die Gitterenthalpie
unterscheiden sich qualitativ. Die Gitterenergie ist eine innere Energie,
während die Gitterenthalpie eine Enthalpie ist. Die Gitterenthalpie
berücksichtigt also zusätzlich die zu leistende Volumenarbeit  gegen einen konstanten äußeren Druck. Hat man für das Auseinanderbringen der
Bestandteile des Festkörpers eine molare Gitterenthalpie
gegen einen konstanten äußeren Druck. Hat man für das Auseinanderbringen der
Bestandteile des Festkörpers eine molare Gitterenthalpie  bestimmt, so ist die molare Gitterenergie
bestimmt, so ist die molare Gitterenergie  .
Hierbei ist
.
Hierbei ist  die auf die Stoffmenge bezogene Volumenänderung.
die auf die Stoffmenge bezogene Volumenänderung.
Die Gitterenthalpie von NaCl setzt sich wie folgt zusammen:
-
NaCl (fest) Na+ (g) + Cl− (g) −411 kJ/mol 611 kJ/mol −244 kJ/mol Bildungsenthalpien (25 °C)
Gitterenthalpie +778 kJ/mol
Vergleich: Dies ist ungefähr die doppelte notwendige Energie, die bei der stark exothermen Reaktion von Natriummetall und Chlorgas freiwerden würde. Die Bildung gasförmiger Ionen ist also extrem endotherm.
Die Gitterenthalpie ΔH0L hängt von Größe und Ladung der beteiligten Ionen ab und ist bei dieser Art der Definition immer positiv, da das Gitter sonst nicht stabil wäre. Eine sehr hohe Gitterenthalpie weist Aluminiumoxid Al2O3 (Al3+ und O2−) mit 15157 kJ/mol auf. Die hohe Gitterenthalpie wird in aluminothermischen Verfahren ausgenutzt; dazu zählen etwa das aluminothermische Schweißen und die Darstellung von Elementen aus ihren Oxiden und Aluminium mittels Aluminothermie. In letzterem Fall ist die hohe Gitterenthalpie des Aluminiumoxids eine Haupttriebkraft für die Reaktion, da sie sich direkt in der Gibbs-Energie niederschlägt.
Häufig wird die Gitterenergie auch als Reaktionsenthalpie bei der Bildung des festen Salzgitters ausgehend von Ionen in der Gasphase definiert. Wird die Gitterenergie so definiert, so ist der Prozess exotherm und die dazugehörige Enthalpieänderung ist negativ anzugeben. Die Gitterenthalpie von Aluminiumoxid wäre dann beispielsweise −15157 kJ/mol.
Die Gitterenthalpie hängt einerseits von der Größe der beteiligten Ionen ab: Je größer die Ionen, desto kleiner ist die frei werdende Gitterenergie, da die Anziehungskräfte mit zunehmender Entfernung der positiven Kerne von der negativen Elektronenhülle des Bindungspartners abnehmen.
Beispiele: molare Gitterenthalpie der Alkalifluoride bei 25 °C in kJ/mol:
| Name | Formel | Ionenradius
der einwertigen Alkalimetall-Kationen X+ in pm |
Gitterenthalpie in kJ pro mol |
|---|---|---|---|
| Lithiumfluorid | LiF | 74 | 1039 |
| Natriumfluorid | NaF | 102 | 920 |
| Kaliumfluorid | KF | 138 | 816 |
| Rubidiumfluorid | RbF | 149 | 780 |
| Caesiumfluorid | CsF | 170 | 749 |
Andererseits hängt die Gitterenergie von der elektrischen Ladung der beteiligten Ionen ab: Je größer die Ladungen, desto größer sind die Anziehungskräfte und umso größer ist die Gitterenergie.
Beispiele: molare Gitterenthalpie bei 25 °C in kJ pro mol (in den Beispielen ändert sich der Ionenradius nur wenig):
| Name | Formel | Kationen | Anionen | Gitterenthalpie in kJ pro mol |
|---|---|---|---|---|
| Natriumchlorid | NaCl | Na+ | Cl− | 780 |
| Natriumsulfid | Na2S | Na+ | S2− | 2207 |
| Magnesiumchlorid | MgCl2 | Mg2+ | Cl− | 2502 |
| Magnesiumsulfid | MgS | Mg2+ | S2− | 3360 |
Solvatationsenthalpie, Hydratationsenthalpie
Sie gibt an, welche Energie freigesetzt wird, wenn sich gasförmige Ionen an Lösemittel anlagern, also solvatisierte Ionen bilden. Für den häufigsten Fall Solvens = Wasser spricht man von Hydratationsenthalpie.
-
Na+ (g) Na+ (hydratisiert) 611 kJ/mol −240 kJ/mol Bildungsenthalpien (25 °C)
Hydratationsenthalpie Na+ −851 kJ/mol (d.i. extrem
exotherm)Cl− (g) Cl− (hydratisiert) −244 kJ/mol −167 kJ/mol Bildungsenthalpien (25 °C)
Hydratationsenthalpie Cl− +77 kJ/mol (d.i. schwach
endotherm)
Die Hydratationsenthalpie der gasförmigen Ionen von Kochsalz ist mit −774 kJ/mol insgesamt stark exotherm.
Lösungsenthalpie / Kristallisationsenthalpie
Die Lösungsenthalpie von Salzen beinhaltet 1) das Separieren des Ionen-Gitters in Einzel-Ionen und 2) die Solvatisierung der Einzel-Ionen. Teilschritt 1) ist sehr stark endotherm, Teilschritt 2) sehr stark exotherm.
- Lösungsenthalpie = Gitterenthalpie + Solvatationsenthalpie.
Lösungsenthalpie NaCl in Wasser = (+778 kJ/mol) + (−851+77 kJ/mol) = +4 kJ/mol (25 °C).
Dieser Wert steht in guter Übereinstimmung mit Tabellenwerken +3,89 kJ/mol für die Kochsalz-Lösungswärme. Beim Lösen tritt also eine ganz geringe Abkühlung der Lösung auf.
Natürlich geht man zur praktischen Berechnungen der Lösungswärme nicht den Umweg über die Gitterenergie, sondern man rechnet direkt und mit nur wenigen Tabellenwerten (gelegentlich findet man den Wert (NaCl)hydrat. = −407 kJ/mol anstelle der Einzel-Ionen):
-
NaCl (s) NaCl (hydratisiert) −411 kJ/mol −407 kJ/mol Bildungsenthalpien (25 °C) NaCl (s) Na+ (hydratisiert) + Cl− (hydratisiert) −411 kJ/mol −240 kJ/mol −167 kJ/mol Bildungsenthalpien (25 °C)
Lösungsenthalpie +4 kJ/mol (Wasser, 25 °C)
Die Solvatationsenthalpie kann immer dann aus den thermodynamischen Daten abgeleitet werden, wenn man Standardbildungsenthalpie-Werte für den festen und gelösten Aggregatzustand findet, z.B. Ameisensäure und Kohlendioxid in obenstehender Tabelle. Sie gilt für „unendliche Verdünnung“.
Bei Umkehrung dieses Prozesses, der Kristallisation aus Lösung, geben die gelösten Ionen eines Salzes 1) ihre Solvathülle ab und 2) schließen sich in einem festen Kristallgitter zusammen. Während des Ausscheidens von Kochsalzkristallen aus Wasser werden −3,89 kJ/mol Kristallisationsenthalpie freigesetzt.
Intermolekulare Enthalpie-Beiträge
Unterschiedlich starke Wechselwirkungen zwischen den Molekülen sind die Ursache dafür, dass Substanzgruppen ähnliche oder stark unterschiedliche Sublimationsenthalpien aufweisen.
- Schwache Beiträge liefern London-Kräfte, Dipol-Dipol-Wechselwirkungen und Ion-Dipol-Wechselwirkungen mit 1–15 kJ/mol Bindung. Sie werden als Van-der-Waals-Wechselwirkung zusammengefasst. Siehe hierzu als Beispiele die Schmelz- und Verdampfungsenthalpien von Wasserstoff, Kohlenmonoxid und Methan.
- Stärkere Beiträge liefern Wasserstoffbrücken-Bindungen mit 20–40 kJ/mol Bindung (je nach Polarisation). Siehe hierzu als Beispiele die Schmelz- und Verdampfungsenthalpien von Wasser, Methanol und Ameisensäure und auch Hydratationsenthalpien. Wasserstoffbrücken-Bindungen sind auch verantwortlich dafür, dass der Siedepunkt von Wasser bei 100 °C liegt, während der von Schwefelwasserstoff nur −83 °C beträgt (siehe Siedepunktanomalie).
- Sehr starke Beiträge liefern Ion-Ion-Wechselwirkungen in Kristallen. Natriumchlorid besteht im Kristall nicht aus diskreten NaCl-Molekülen, sondern aus einer gleichen Anzahl von Natriumkationen und Chloridanionen, die sich im Kristallgitter entsprechend den Coulomb-Kräften exakt angeordnet haben.
Enthalpie in der Chemie (Thermochemie)
Die Thermochemie beschreibt im engeren Sinne nur die intramolekularen Kräfte, also die energetischen Beziehungen zwischen den einzelnen Atomen eines Moleküls. Kovalente Bindungen beinhalten ca. 150–1000 kJ/mol Bindungsenergie, ionische Bindungen ca. fünfmal so große Beträge.
Bei Kenntnis der Standardbildungsenthalpien von Edukten und Produkten lässt sich eine mögliche chemische Reaktion energetisch grob bilanzieren. Die wichtigste Frage ist oft, ob ein Prozess endotherm oder exotherm verläuft und wie stark.
Durch Details wie Verdampfungs-, Schmelz-, Solvatations- oder Kristallisationsenthalpien können Teilschritte innerhalb der chem. Reaktion energetisch präzisiert werden.
Reaktionsenthalpie
Die Reaktionsenthalpie ist diejenige Energie, die freigesetzt oder benötigt wird, wenn zwischen den Molekülen zweier Stoffe neue chemische Bindungen gebildet werden. Sie ist abhängig von den Reaktionspartnern (Edukte) und der Art der chemischen Bindung im Produkt. Zur Berechnung vergleicht man die Summe der Bildungsenthalpien der Produkte mit der der Edukte. Die Differenz ist die Reaktionsenthalpie, die anschließend durch Bezug auf die Stoffmenge des jeweils interessierenden Produkts standardisiert werden kann:
-
2 Na (s) + Cl2 (g) 2 NaCl (s) 2 × 0 kJ/mol 0 kJ/mol 2 × −411 kJ/mol Bildungsenthalpien (25 °C)
Reaktionsenthalpie = 2 mol × (−411) kJ/mol − 2 mol × 0 kJ/mol − 1 mol ×
0 kJ/mol = −822 kJ.
- Also verläuft die Reaktion exotherm. Division durch die erhaltene Stoffmenge, in diesem Fall 2 Mol Natriumchlorid, liefert dessen (in diesem Beispiel allerdings schon zu Beginn vorausgesetzte) molare Bildungsenthalpie von −411 kJ/mol NaCl (25 °C).
Standardverbrennungsenthalpie
Auch die Verbrennung ist eine chemische Reaktion. Die Reaktionsenthalpie der Verbrennungsreaktion bzw. die Standardverbrennungsenthalpie eines Stoffes ist die Enthalpieänderung, die auftritt, wenn ein Stoff unter O2-Überschuss (O2-Überdruck) und Standardbedingungen (101,325 kPa und 25 °C) vollständig verbrennt. Definitionsgemäß bezieht sich diese Verbrennungswärme auf die Bildung von gasförmigem Kohlendioxid und flüssigem Wasser (bzw. N2) als Endprodukte; unter Sauerstoffüberdruck kann sich kein gasförmiges Wasser bilden. Sie wird mit ΔVH0 bezeichnet und ihr absoluter Betrag ist identisch mit dem Brennwert Hs.
In einem Autoklaven-Rohr wird folgende Reaktion mit Sauerstoffüberdruck durchgeführt:
-
C3H8 (g) + 5 O2 (g) 3 CO2 (g) + 4 H2O (l) −103,2 kJ/mol 5 × 0 kJ/mol 3 × −393,5 kJ/mol 4 × −285,8 kJ/mol
-
Reaktionsenthalpie = 3 mol × (−393,5) kJ/mol + 4 mol × (−285,8) kJ/mol – 1 mol
× (−103,2) kJ/mol − 5 mol × (0) kJ/mol = -2,22 MJ.
- Division durch die eingesetzte Stoffmenge, in diesem Fall 1 Mol Propan, liefert dessen molare Verbrennungsenthalpie von −2,22 MJ/mol Propan (25 °C)
Die gleiche Reaktion in einer offenen Brennerflamme; es entstehen nur gasförmige Verbrennungsprodukte:
-
C3H8 (g) + 5 O2 (g) 3 CO2 (g) + 4 H2O (g) −103,2 kJ/mol 5 × 0 kJ/mol 3 × −393,5 kJ/mol 4 × −241,8 kJ/mol
-
Reaktionsenthalpie = 3 mol × (−393,5) kJ/mol + 4 mol × (−241,8) kJ/mol – 1 mol
× (−103,2) kJ/mol − 5 mol × (0) kJ/mol = -2,04 MJ.
- Division durch die eingesetzte Stoffmenge, in diesem Fall 1 Mol Propan, liefert dessen molare Verbrennungsenthalpie von −2,04 MJ/mol Propan (25 °C)
Fortgeschrittene Anwendung
Es ist müßig, sich für jede Reaktion die Standardbildungsenthalpien der Edukte und Produkte zusammenzusuchen, zudem noch im korrekten Aggregatzustand. Zudem stößt man bei größeren Molekülen schnell in ein „Datenvakuum“. Folgende vereinfachte Betrachtungen haben sich in der Praxis bewährt:
- Es ist unerheblich, ob man ein langkettig alkylsubstituiertes Ethylen bromiert oder Ethylen selbst, die Reaktionswärme pro (C=C)-Doppelbindung ist weitgehend gleich.
- Es ist unerheblich, ob man eine Reaktion komplett in flüssiger Phase berechnet oder komplett in der Gasphase, die Reaktionswärme beeinflusst dies kaum.
- Es ist unerheblich (einige Abweichungen), wenn man bei 150 °C durchgeführte Reaktionen für 25 °C Standardbedingungen berechnet. (Die Reaktionsenthalpie kann für beliebige Temperaturen berechnet werden bei Kenntnis der Temperaturabhängigkeit der Molwärmen aller Reaktionspartner.)
Daher kann man normale organisch-chemische Umsetzungen wie Halogen-Additionen, Cycloadditionen, Veresterungen mit Säuren oder Anhydriden, Hydrolysen etc. mit Hilfe von zahlreich tabellierten Inkrementen für gasförmige Moleküle nach Benson berechnen.
Im nachfolgenden Beispiel wird die Reaktionsenergie der Brom-Addition an Ethylen mit Benson-Inkrementen berechnet und zum Vergleich aus Bindungsenergien involvierter Bindungen abgeschätzt. (Merke: Bindungsenergien sind gemittelte Dissoziationsenergien, keine Standardbildungsenthalpien!)
Bromaddition an ein Alken, Reaktionsenthalpie berechnet mit Standardbildungsenthalpien:
-
H2C=CH2 + Br2 H2BrC-CBrH2 52 kJ/mol 0 kJ/mol −39 kJ/mol
-
Reaktionsenthalpie (25 °C, alle Werte für gasförm. Zustand) = (−39 − 52)
kJ/mol = −91 kJ/mol Doppelbindung
Bromaddition an ein Alken, Reaktionsenthalpie berechnet mit Inkrementen nach Benson:
-
H2C=CH2 + Br2 H2BrC-CBrH2 2 Cd-(H)2: 2× +28,1 kJ/mol 2 C-(H)2(Br)(C): 2× −22,6 kJ/mol
-
Reaktionsenthalpie (berechnet nach Benson)
= ( −45,2 − (+56,2)) kJ/mol = −101 kJ/mol Doppelbindung
Bromaddition an ein Alken, Reaktionsenthalpie abgeschätzt mit Bindungsenergien:
-
H2C=CH2 + Br2 H2BrC-CBrH2 4 C-H: 4× > 455 kJ/mol 4 C-H: 4× 380±50 kJ/mol 1 C=C: 1× 614 kJ/mol 1 C-C: 1× 348 kJ/mol 1 Br-Br: 193 kJ/mol 2 C-Br: 2× 260±30 kJ/mol
-
Reaktionsenthalpie (geschätzt aus Bindungsenergien) = ( 2388±200 - 2627)
kJ/mol = -239±200 kJ/mol Doppelbindung
Die beste Abschätzung für Reaktionsenthalpien gelingt mit Standardbildungsenthalpien oder Inkrementen nach Benson, bei Verwendung von „Bindungsenergien“ ist die Unsicherheit mit ±200 kJ/mol viel zu hoch.
Technische Anwendbarkeit
Die Reaktionsenthalpien organischer Reaktionen liegen im Bereich −160 bis +100 kJ pro mol „reaktiver Zentren“. Als sehr stark exotherm erweisen sich alle Additionsreaktionen mit Epoxiden, Anhydriden und Halogenen. Diese thermochemischen Betrachtungen treffen keine Aussage darüber, wie schnell diese Reaktionswärmen freigesetzt werden. Sie machen nur die Aussage, dass bis zum Reaktionsende diese Wärme freigesetzt wird. Jede Reaktion erhöht ihre Geschwindigkeit um das Zwei- bis Dreifache bei Temperaturerhöhung um 10 K. Umgekehrt bedeutet eine zweifache Verdünnung der Reaktionspartner häufig eine Halbierung der Reaktionsgeschwindigkeit bzw. Wärmeleistung der Reaktion. Berechnete Reaktionsenthalpien dienen dazu, in einem System von Reaktanten und Lösemittel über deren Wärmekapazitäten den Temperaturverlauf zu berechnen. Großtechnische Anlagen verfügen nur über begrenzte Kühlkapazitäten (-Wärme/Zeit), dies bleibt im Laborversuch häufig wenig berücksichtigt.
Bindungsenergie / Dissoziationsenergie
Die Bindungsenergie bzw. Bindungsstärke gibt die „Stabilität“ der kovalenten Bindung an. Die Bestimmung ist nur bei zweiatomig symmetrischen Molekülen wie z.B. Wasserstoff oder Halogene direkt möglich. In diesen Fällen kann die Dissoziationsenergie zur Bildung zweier identischer Radikale einfach gemessen/berechnet werden. Bei „Element-Radikalen“ bezeichnet man die Standardbildungsenthalpie von Radikalen auch als Atomisierungsenthalpien.
In allen anderen Fällen sind Werte für die „Bindungsenergie“ nur indirekt möglich durch Vergleich mehrerer Dissoziationsenergie-Messungen an homologen Molekülen. Die Werte schwanken abhängig vom Substitutionsmuster an den Radikal-Zentren.
Die Standardbildungsenthalpie von gasförmigen
- Brom-Radikalen beinhaltet die Verdampfungsenthalpie (31 kJ/mol), die notwendig ist, um flüssiges Brom in gasförmiges Brom zu verwandeln.
- Iod-Radikalen beinhaltet die Sublimationsenthalpie (62 kJ/mol), die notwendig ist, um kristallines Iod in gasförmiges Iod zu verwandeln.
- Kohlenstoff-Radikalen ist identisch mit der Standardbildungsenthalpie von gasförmigem Kohlenstoffdampf.
Thermodynamische Eigenschaften der Enthalpie
Enthalpie als Fundamentalfunktion
Betrachtet man ein System, dessen Eigenschaften durch die Zustandsgrößen
Entropie ,
Volumen
und Molzahlen  der
der  chemischen Komponenten gegeben sind, dann ist die innere Energie
des Systems, ausgedrückt als Funktion der genannten Zustandsgrößen (nämlich
aller extensiven Variablen des Systems),
chemischen Komponenten gegeben sind, dann ist die innere Energie
des Systems, ausgedrückt als Funktion der genannten Zustandsgrößen (nämlich
aller extensiven Variablen des Systems),

eine Fundamentalfunktion des Systems. Sie beschreibt das System vollständig, es lassen sich alle thermodynamischen Eigenschaften des Systems aus ihr ableiten.
Oft sind diese Variablen jedoch für die praktische Arbeit ungünstig und man
würde vorziehen, etwa die Temperatur oder den Druck in der Variablenliste zu
haben. Im Gegensatz zur sonst üblichen Vorgehensweise darf ein Variablenwechsel
im vorliegenden Fall jedoch nicht durch eine einfache Substitution geschehen, da
sonst Information verloren geht. Soll beispielsweise das Volumen durch den Druck
ersetzt werden, könnte
aus den Funktionen  und
und  eliminiert werden, um eine Funktion der Form
eliminiert werden, um eine Funktion der Form  zu erhalten. Da jedoch der Druck thermodynamisch als partielle Ableitung der
inneren Energie nach dem Volumen definiert ist
zu erhalten. Da jedoch der Druck thermodynamisch als partielle Ableitung der
inneren Energie nach dem Volumen definiert ist

wäre diese Formulierung gleichbedeutend mit einer partiellen
Differentialgleichung für ,
welche
nur bis auf unbestimmte Funktionen festlegen würde. Dieses
wäre nach wie vor eine Beschreibung des betrachteten Systems, aber es wäre keine
vollständige Beschreibung und damit keine Fundamentalfunktion mehr.
Zum Variablenwechsel unter Erhaltung der vollständigen Information muss eine
Legendre-Transformation
durchgeführt werden. Soll beispielsweise zur Variablenliste  übergegangen werden, lautet die Transformation:
übergegangen werden, lautet die Transformation:
 .
.
Die Legendre-Transformierte  wird Enthalpie genannt. Sie ist wiederum eine Fundamentalfunktion,
wenn sie als Funktion der Variablen
– dies sind die natürlichen Variablen der Enthalpie – gegeben ist. Sie
kann auch in Abhängigkeit von anderen Variablen ausgedrückt werden, ist dann
aber keine Fundamentalfunktion mehr.
wird Enthalpie genannt. Sie ist wiederum eine Fundamentalfunktion,
wenn sie als Funktion der Variablen
– dies sind die natürlichen Variablen der Enthalpie – gegeben ist. Sie
kann auch in Abhängigkeit von anderen Variablen ausgedrückt werden, ist dann
aber keine Fundamentalfunktion mehr.
Die Herkunft der Enthalpie aus einer Legendre-Transformation erklärt den
additiven Term  :
Er kompensiert den Informationsverlust, der sonst mit dem Variablenwechsel
verbunden wäre.
:
Er kompensiert den Informationsverlust, der sonst mit dem Variablenwechsel
verbunden wäre.
Fundamentalfunktionen, welche die Dimension Energie besitzen, heißen auch thermodynamische Potentiale. Die Enthalpie ist also ein thermodynamisches Potential.
Ableitungen der Enthalpie
Geht man von der inneren Energie als Funktion ihrer natürlichen Variablen aus und bildet ihr totales Differential, erhält man:
 .
.
Die hierbei auftretenden partiellen Ableitungen werden in der Thermodynamik
als die Definitionen von Temperatur  ,
Druck
und chemischem
Potential der i-ten Substanz
,
Druck
und chemischem
Potential der i-ten Substanz  interpretiert:
interpretiert:

so dass sich das Differential auch schreiben lässt als
 .
.
Das totale Differential der Enthalpie als Funktion ihrer natürlichen Variablen ist einerseits formal
 .
.
und andererseits, unter Benutzung ihrer Definition

so dass aus dem Vergleich der Koeffizienten in den markierten Gleichungen folgt
 ,
,
sowie

und
 .
.
Die Herleitung zeigt gleichzeitig, wie die Addition des Terms
die Liste der unabhängigen Variablen von  in
in  ändert,
indem dadurch im totalen Differential der von
abhängige Term entfernt und ein von
abhängiger Term hinzugefügt wird.
ändert,
indem dadurch im totalen Differential der von
abhängige Term entfernt und ein von
abhängiger Term hinzugefügt wird.
Die zweite der markierten Gleichungen ist eine „differentielle Fundamentalfunktion“, nämlich die differentielle Enthalpie als Funktion ihrer natürlichen Variablen:
 .
.
Minimumsprinzip der Enthalpie
Gemäß dem Zweiten Hauptsatz der Thermodynamik nimmt ein abgeschlossenes System unter den erreichbaren Zuständen denjenigen als Gleichgewichtszustand ein, der bei der gegebenen inneren Energie die höchste Entropie besitzt. Aus diesem Maximumsprinzip der Entropie lässt sich ein Minimumsprinzip der inneren Energie ableiten: Bei konstant gehaltener Entropie nimmt ein System denjenigen Zustand als Gleichgewichtszustand ein, der die geringste innere Energie besitzt.
Ein ähnliches Minimumsprinzip existiert für die Enthalpie: Ein System, das auf konstantem Druck gehalten wird, nimmt von allen erreichbaren Zuständen mit diesem Druck denjenigen als Gleichgewichtszustand ein, in dem die Enthalpie den kleinstmöglichen Wert hat.
Zum Beweis
betrachte man ein System, dessen Druck auf einem konstanten Wert gehalten wird.
Die Druckregelung kann beispielsweise dadurch geschehen, dass das betrachtete
System über eine bewegliche adiabatische Wand in Kontakt mit einem zweiten
System steht, das unveränderlich den gewünschten Druck aufweist (in
thermodynamischer Ausdrucksweise: ein Druckreservoir). Durch Verschiebung der
Kontaktwand kann das betrachtete System bei Bedarf so lange mit dem
Druckreservoir „Volumen austauschen“, bis es seinen Druck wieder dem des
Reservoirs angeglichen hat. Das aus dem betrachteten System und dem
Druckreservoir gebildete Gesamtsystem nimmt bei konstant gehaltener Entropie
gemäß dem Energieminimums-Prinzip die geringstmögliche innere Energie  an, und im Energieminimum gilt:
an, und im Energieminimum gilt:
 .
.
Da mit dem Reservoir gemäß dessen Definition ausschließlich Volumen
ausgetauscht wird, kann die innere Energie des Reservoirs nur dadurch geändert
werden, dass Volumenänderungsarbeit an ihm geleistet wird:  ,
und damit ist
,
und damit ist
 .
.
Bei der Verschiebung der Trennwand gilt  ,
so dass
,
so dass
 .
.
Da der Druck des Reservoirs,  ,
gemäß Voraussetzung konstant ist, lässt sich schreiben
,
gemäß Voraussetzung konstant ist, lässt sich schreiben
 .
.
Es wären nun die Extremaleigenschaften der Funktion  näher zu untersuchen. Da man jedoch Druckausgleich zwischen dem betrachteten
System (Druck )
und dem Druckreservoir (Druck )
erwartet, ist es naheliegend, die weitere Untersuchung auf die Zustände mit der
Eigenschaft
näher zu untersuchen. Da man jedoch Druckausgleich zwischen dem betrachteten
System (Druck )
und dem Druckreservoir (Druck )
erwartet, ist es naheliegend, die weitere Untersuchung auf die Zustände mit der
Eigenschaft  zu beschränken. Auf der Untermenge dieser Zustände ist aber
identisch mit
zu beschränken. Auf der Untermenge dieser Zustände ist aber
identisch mit  .
Es gilt also
.
Es gilt also
 ,
unter der Voraussetzung .
,
unter der Voraussetzung .
Wie sich zeigen lässt,
ist die zweite Ableitung von
in diesem Zustand positiv, so dass das gefundene Extremum der Enthalpie
tatsächlich ein Minimum ist.
Das Minimumsprinzip für die innere Energie des Gesamtsystems bei
konstanter Entropie führt also dazu, dass die Enthalpie des betrachteten
Systems auf der Untermenge der Zustände mit konstantem Druck
ein Minimum annimmt. Ist das System noch nicht im Gleichgewicht, bewegt es sich
(unter isobaren Bedingungen) freiwillig in Zustände niedrigerer Enthalpie. Das
Gleichgewicht ist mit dem Zustand erreicht, in dem die Enthalpie den unter den
gegebenen Bedingungen kleinstmöglichen Wert besitzt.
Will man den Gleichgewichtszustand mit Hilfe des (allgemein und stets gültigen) Entropiekriteriums bestimmen, muss das Maximum der Gesamtentropie ermittelt werden, also die Summe der Entropien des untersuchten Systems und seiner Umgebung. Es muss daher nicht nur die Änderung der System-Entropie bei einer Zustandsänderung betrachtet werden, sondern auch die Entropie-Änderung, die das System durch Rückwirkung auf die Umgebung dort erzeugt. Das Enthalpiekriterium ist eine Umformulierung des Entropiekriteriums, in welche ausschließlich Eigenschaften des betrachteten Systems eingehen und welche die Rückwirkung auf die Umgebung (unter isobaren Bedingungen) automatisch berücksichtigt. Bei Verwendung des Enthalpiekriteriums kann die Ermittlung des (isobaren) Gleichgewichtszustands sich also auf die Betrachtung des Systems beschränken, was die Untersuchungen merklich erleichtert.
Wird die Enthalpie als Funktion ihrer natürlichen Variablen  ausgedrückt, reduziert die Bedingung konstanten Drucks die Liste der Variablen
um
und die Enthalpie hängt nur noch von den Variablen
ausgedrückt, reduziert die Bedingung konstanten Drucks die Liste der Variablen
um
und die Enthalpie hängt nur noch von den Variablen  ab. Diese Vereinfachung demonstriert einen der möglichen Vorteile bei der
Verwendung der Enthalpie in geeigneten Fällen. Der Gleichgewichtszustand könnte
auch durch Aufsuchen des Minimums der inneren Energie des betrachteten Systems,
,
ermittelt werden, aber die Bedingung konstanten Drucks würde in diesem Fall
nicht zu einer Vereinfachung der Funktion führen, sondern zu komplizierten
Abhängigkeiten zwischen den Variablen
ab. Diese Vereinfachung demonstriert einen der möglichen Vorteile bei der
Verwendung der Enthalpie in geeigneten Fällen. Der Gleichgewichtszustand könnte
auch durch Aufsuchen des Minimums der inneren Energie des betrachteten Systems,
,
ermittelt werden, aber die Bedingung konstanten Drucks würde in diesem Fall
nicht zu einer Vereinfachung der Funktion führen, sondern zu komplizierten
Abhängigkeiten zwischen den Variablen  .
Zusätzliche Probleme würden dabei entstehen, falls die Zustandsgleichung
.
Zusätzliche Probleme würden dabei entstehen, falls die Zustandsgleichung  nicht explizit bekannt ist.
nicht explizit bekannt ist.
Für einen realen physikalischen oder chemischen Prozess kann oft die Atmosphäre als Druckreservoir dienen. Wegen ihres großen Volumens ändert sich ihr Druck nicht nennenswert, wenn ein System Volumenänderungsarbeit an ihr leistet. Die Voraussetzungen für die Anwendbarkeit des Minimumsprinzips der Enthalpie sind also erfüllt, wenn ein System adiabatisch gegen die Umwelt isoliert ist (um Wärmeaustausch mit der Umgebung zu verhindern und damit die Entropie konstant zu halten) und über einen beweglichen Kolben oder eine ähnliche Vorrichtung an den atmosphärischen Umgebungsdruck gekoppelt ist (um den Systemdruck konstant zu halten).
In der Laborpraxis kommen solche adiabatischen Systeme allerdings selten vor.
Chemische Reaktionen finden meistens nicht in adiabatisch isolierten Gefäßen
statt, so dass Wärme mit der Umgebung ausgetauscht werden kann. Die Atmosphäre
dient dann nicht nur als Druck-, sondern auch als Wärmereservoir:
Sie hält Druck und Temperatur konstant. Das thermodynamische Potential,
das unter diesen Bedingungen ein Minimum annimmt, ist die Gibbs-Energie
 .
.
Enthalpie und Wärmekapazität
Führt man einem System die spezifische (d.h. auf die Masse bezogene)
Wärmemenge  zu und bewirkt dadurch eine Temperaturänderung
zu und bewirkt dadurch eine Temperaturänderung  ,
dann ist die spezifische
Wärmekapazität
,
dann ist die spezifische
Wärmekapazität  des Systems definiert durch die Gleichung
des Systems definiert durch die Gleichung

oder
 .
.
Die spezifische Wärmekapazität ist nicht nur vom Material, sondern auch von
der Prozessführung abhängig. Erfolgt die Wärmezufuhr isochor
(d.h. bei konstantem Volumen), dann trägt die gesamte zugeführte
Wärmemenge zur Erhöhung der spezifischen inneren Energie  bei:
bei:

und der Prozess wird beschrieben durch die spezifische Wärmekapazität bei
konstantem Volumen, 
 ,
,
welche also die Ableitung der spezifischen inneren Energie nach der Temperatur bei konstantem Volumen ist.
Erfolgt die Wärmezufuhr isobar, dann ist die zugeführte spezifische
Wärmemenge gleich der Erhöhung der spezifischen Enthalpie  :
:

und der Prozess wird beschrieben durch die spezifische Wärmekapazität bei
konstantem Druck,
 ,
,
welche also die Ableitung der spezifischen Enthalpie nach der Temperatur bei konstantem Druck ist.
Da zur Erzielung einer gewünschten Temperaturerhöhung im isobaren Fall die vom System zu leistende Volumenänderungsarbeit zusätzlich zugeführt werden muss, ist zu erwarten, dass die isobare spezifische Wärmekapazität einer Substanz größer sein wird als die isochore. Eine genauere Betrachtung muss berücksichtigen, dass die Ausdehnung des Systems meistens auch dessen innere Energie verändert. Es lässt sich jedoch zeigen, dass allgemein gilt:
 .
.
Auf der rechten Seite können die Temperatur ,
das spezifische Volumen  ,
das Quadrat des isobaren thermischen Ausdehnungskoeffizienten
,
das Quadrat des isobaren thermischen Ausdehnungskoeffizienten
 und (aus thermodynamischen Stabilitätsgründen) die isotherme Kompressibilität
und (aus thermodynamischen Stabilitätsgründen) die isotherme Kompressibilität
 nicht negativ werden, so dass stets
nicht negativ werden, so dass stets

ist. In manchen Fällen kann
um 30 % größer sein als .
Temperatur- und Druckabhängigkeit der Enthalpie
Tabellierungen der Enthalpie beziehen sich aus Platzgründen in der Regel auf eine bestimmte Temperatur und einen bestimmten Druck. Soll die Enthalpie für andere Bedingungen ermittelt werden, sind Formeln wünschenswert, die den Übergang vom Referenzzustand auf andere Temperaturen und Drücke erlauben. Es ist vorteilhaft, wenn für die Umrechnung nur die Kenntnis direkt messbarer Größen benötigt wird.
Für ein geschlossenes System, das sich im Gleichgewicht befindet und in dem
keine chemischen Reaktionen stattfinden ( )
ist, wie oben ausgeführt, eine infinitesimale Änderung der Enthalpie gegeben
durch
)
ist, wie oben ausgeführt, eine infinitesimale Änderung der Enthalpie gegeben
durch
 .
.
Das Differential  lässt sich – aufgefasst als Funktion der hier interessierenden Variablen
und
– wie folgt entwickeln:
lässt sich – aufgefasst als Funktion der hier interessierenden Variablen
und
– wie folgt entwickeln:

Mit Hilfe der Identitäten
 (wegen
(wegen  gemäß dem Zweiten
Hauptsatz)
gemäß dem Zweiten
Hauptsatz)
und
 (einer der Maxwell-Beziehungen)
(einer der Maxwell-Beziehungen)
folgt
 ,
,
was sich unter Benutzung der isobaren Wärmekapazität  und des isobaren thermischen Ausdehnungskoeffizienten
und des isobaren thermischen Ausdehnungskoeffizienten  ausschließlich durch die messbaren Größen ,
ausschließlich durch die messbaren Größen ,
 und
ausdrücken lässt:
und
ausdrücken lässt:
 .
.
Für eine endlich große Zustandsänderung vom Zustand  in den Zustand
in den Zustand  ist über diese infinitesimale Zustandsänderung zu integrieren:
ist über diese infinitesimale Zustandsänderung zu integrieren:
 .
.
Falls die Zustandsänderung mit einem Phasenübergang verbunden ist, muss die betreffende Latentwärme zusätzlich berücksichtigt werden.
Die Größen ,
und
sind in der Regel selbst abhängig von Temperatur und Druck. Diese Abhängigkeiten
müssen bekannt sein, damit die Integrale ausgeführt werden können.
Da die Enthalpie eine Zustandsgröße ist, hängen die Integrale nicht von der
Wahl des Pfades ab, entlang welchem integriert wird. Eine bequeme Wahl des
Pfades besteht darin, zunächst das Temperaturintegral auf einem Pfad konstanten
Druckes auszuführen, der den Anfangszustand
mit einem Zwischenzustand  verbindet. Dazu muss
im Temperaturbereich von
verbindet. Dazu muss
im Temperaturbereich von  bis
bis  beim fixen Druck
bekannt sein. Anschließend wird das Druckintegral auf dem Pfad konstanter
Temperatur ausgeführt, der
mit
verbindet. Dazu muss
beim fixen Druck
bekannt sein. Anschließend wird das Druckintegral auf dem Pfad konstanter
Temperatur ausgeführt, der
mit
verbindet. Dazu muss  im Druckbereich von
bis
bei der fixen Temperatur
bekannt sein.
im Druckbereich von
bis
bei der fixen Temperatur
bekannt sein.
Alternativ kann die Integration erst auf einem Pfad konstanter Temperatur von
nach  geführt werden, wozu
im Druckbereich von
bis
bei der fixen Temperatur
bekannt sein muss. Anschließend führt der Pfad bei konstantem Druck von
nach ,
wozu
im Temperaturbereich von
bis
beim fixen Druck
bekannt sein muss.
geführt werden, wozu
im Druckbereich von
bis
bei der fixen Temperatur
bekannt sein muss. Anschließend führt der Pfad bei konstantem Druck von
nach ,
wozu
im Temperaturbereich von
bis
beim fixen Druck
bekannt sein muss.
Wird der Prozess isobar geführt, ist  ,
das Druckintegral fällt weg und es ist lediglich die Kenntnis der
temperaturabhängigen Wärmekapazität
beim betreffenden fixen Druck nötig:
,
das Druckintegral fällt weg und es ist lediglich die Kenntnis der
temperaturabhängigen Wärmekapazität
beim betreffenden fixen Druck nötig:
 .
.
Siehe auch
- Thermodynamisches Potential
- Gibbs-Energie
- Isenthalp
- Gruppenbeitragsmethoden (zur Vorhersage von Verdampfungsenthalpien)


© biancahoegel.de
Datum der letzten Änderung: Jena, den: 09.12. 2025