Phosphofructokinase 1
| Phosphofructokinase 1 | ||
|---|---|---|
| ||
| Masse/Länge Primärstruktur | 780 Aminosäuren | |
| Sekundär- bis Quartärstruktur | Tetramer MMMM, LLLL, MMLL etc. | |
| Kofaktor | Mg2+ | |
| Isoformen | L1, L2, M1, M2, P | |
| Bezeichner | ||
| Gen-Name(n) |
| |
| Externe IDs |
| |
| Enzymklassifikation | ||
| EC, Kategorie | ||
| Reaktionsart | Phosphorylierung | |
| Substrat | ATP + D-Fructose-6-Phosphat | |
| Produkte | ADP + D-Fructose-1,6-bisphosphat | |
| Vorkommen | ||
| Übergeordnetes Taxon | Lebewesen | |
Phosphofructokinase 1, abgekürzt PFK1 oder PFK, auch Fructose-6-phosphat-kinase ist ein Enzym, welches den geschwindigkeitsbestimmenden Schritt der Glykolyse katalysiert, die Umwandlung von Fructose-6-phosphat zu Fructose-1,6-bisphosphat. Phosphofructokinase bestimmt entscheidend mit, wie viel verfügbare Energie die Zelle (ATP, Citrat, NADH/H+) besitzt. Sie kommt in allen Lebewesen vor. Im Menschen gibt es fünf Isoformen, die von drei verschiedenen Genen produziert werden: PFKM (Muskel), PFKL (Leber), PFKP (Blutplättchen). Mutationen in PFKM sind die Ursache für die seltene Tarui-Krankheit.[1]
Struktur
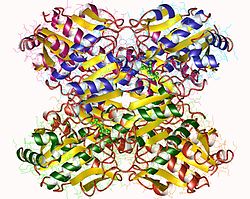
Die PFK1 aus Säugetieren ist ein 340 kDa[2] schweres Tetramer, das aus verschiedenen Kombinationen von drei Arten von Untereinheiten besteht: Muskel (M), Leber (L) und Blutplättchen (P). Die Zusammensetzung des PFK1-Tetramers unterscheidet sich je nach Gewebetyp, in dem es vorliegt. Beispielsweise exprimiert ein reifer Muskel nur das M-Isozym, daher besteht die PFK1 des Muskels ausschließlich aus Homotetrameren von M4. Leber und Nieren exprimieren überwiegend die L-Isoform. In Erythrozyten tetramerisieren sowohl M- als auch L-Untereinheiten zufällig, um M4 sowie L4 und die drei Hybridformen des Enzyms (ML3, M2L2, M3L) zu bilden. Infolgedessen hängen die kinetischen und regulatorischen Eigenschaften der verschiedenen Isoenzyme von der Zusammensetzung der Untereinheiten ab. Gewebespezifische Veränderungen der PFK-Aktivität und des isoenzymischen Gehalts tragen erheblich zur Vielfältigkeit der Glykolyse- und Gluconeogenese-Raten bei, die für verschiedene Gewebe beobachtet wurden.[3]
PFK1 ist ein allosterisches Enzym und hat eine ähnliche Struktur wie Hämoglobin, sofern es sich um ein Dimer eines Dimers handelt.[4] Jede Untereinheit des Tetramers besteht aus 319 Aminosäuren und besteht aus zwei Domänen. Eine Domäne enthält die ATP-Bindungsstelle, die andere Hälfte die Substratbindungsstelle (Fructose-6-phosphat, F6P) sowie eine separate allosterische Bindungsstelle.[5] Jede Domäne ist ein β-Fass und hat ein zylindrisches β-Faltblatt, das von α-Helices umgeben ist.
Auf der gegenüberliegenden Seite jeder Untereinheit von jedem aktiven Zentrum befindet sich die allosterische Stelle an der Grenzfläche zwischen den Untereinheiten im Dimer. ATP und AMP konkurrieren um diese Bindungsstelle. Die N-terminale Domäne hat eine katalytische Rolle, die das ATP bindet und die C-terminale Domäne eine regulatorische Rolle.[6]
Mechanismus
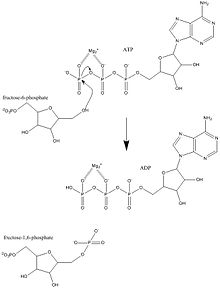
Fructose-6-phosphat wirkt als allosterischer Aktivator. PFK1 bevorzugt das β-Anomer des Fructose-6-phosphats. Fructose-6-phosphat induziert beim Binden eine reaktive Konformation, die sowohl sich selbst als auch ATP für den basenkatalysierten Phosphattransfer orientiert. Fructose-6-phosphat wird bei der Deprotonierung der Hydroxygruppe am C1-Atom durch Asp128 als Nucleophil aktiviert. Das Sauerstoff-Nucleophil entzieht ATP das γ-Phosphat und bildet ADP. Anschließend wird Asp128 deprotoniert. Kinetische Studien deuten darauf hin, dass Asp128 für die Rückreaktion protoniert werden muss. Kristallstrukturen zeigen ein Wassermolekül im aktiven Zentrum. Dies soll den Protonentransfer von und zu Asp128 erleichtern.[7]
PFK1 ist ein allosterisches Enzym, dessen Aktivität mit dem Symmetrie-Modell der Allosterie[8] beschrieben werden kann, wobei ein konzertierter Übergang von einem enzymatisch inaktiven T-Zustand (tense state) in den aktiven R-Zustand (relaxed state) erfolgt. F6P bindet mit hoher Affinität an das Enzym im R-Zustand, jedoch nicht an das Enzym im T-Zustand. Für jedes F6P-Molekül, das an PFK1 bindet, wechselt das Enzym progressiv vom T-Zustand in den R-Zustand. Ein Graph, in dem die PFK1-Aktivität gegen steigende F6P-Konzentrationen aufgetragen ist, würde somit den sigmoiden Kurvenverlauf annehmen, der traditionell mit allosterischen Enzymen assoziiert ist.
PFK1 gehört zur Familie der Phosphotransferasen und katalysiert den Transfer von γ-Phosphat von ATP zu Fructose-6-phosphat. Das aktive Zentrum umfasst sowohl ATP, Mg2+ als auch die F6P-Bindungsstellen. Allosterische Aktivatoren wie AMP und ADP binden an die allosterische Stelle, um die Bildung des R-Zustands durch Induzieren von Strukturänderungen im Enzym zu erleichtern. In ähnlicher Weise binden Inhibitoren wie ATP und PEP an dieselbe allosterische Stelle und erleichtern die Bildung des T-Zustands, wodurch die Enzymaktivität inhibiert wird.
Einige vorgeschlagene Reste, die an der Substratbindung von PFK1 in E. coli beteiligt sind, umfassen Asp127 und Arg171.[9]
In der PFK1 von Geobacillus stearothermophilus bildet die positiv geladene Seitenkette des Arg162-Restes eine Wasserstoff-gebundene Salzbrücke mit der negativ geladenen Phosphatgruppe von F6P, eine Wechselwirkung, die den R-Zustand gegenüber den T-Zustand stabilisiert und teilweise für den homotropen Effekt der F6P-Bindung verantwortlich ist. Im T-Zustand verschiebt sich die Enzymkonformation geringfügig, so dass der zuvor vom Arg162 eingenommene Raum durch Glu161 ersetzt wird. Dieses Vertauschen von Positionen zwischen benachbarten Aminosäureresten hemmt die Fähigkeit von F6P, das Enzym zu binden.
Stellung im Energiestoffwechsel
Die zentrale Rolle der Glykolyse im Energiestoffwechsel begründet sich darin, dass das Substrat für diesen Prozess aus ganz unterschiedlichen Abbauwegen stammt, die damit zusammengeführt werden. Der weitere Abbau in der Glycolyse setzt letztlich Energie frei, die genutzt wird, um energiereiches ATP zu synthetisieren. Der Umsatz in der Glycolyse wird reguliert, indem die beteiligten Enzyme gehemmt werden. Denkbar hierfür wären neben der Reaktion der Phosphofructokinase (PFK) auch die der Pyruvatkinase und andere. Als wichtigste Kontrollstelle gilt aber heute die PFK, die die Umwandlung von Fructose-6-phosphat in Fructose-1,6-bisphosphat katalysiert. PFK wird durch höhere ATP-Konzentrationen inhibiert, betrachtet man die Glycolyse als Ganzes, so lässt sich dies als Endprodukthemmung der Glycolyse deuten. Auf der Ebene der Enzymkinetik bedeutet die Hemmung durch ATP, dass die Michaelis-Menten-Konstante (Km-Wert) der PFK durch ATP steigt.
Diese Eigenschaften der PFK bilden den wichtigsten Aspekt molekularer Erklärungen des Pasteur-Effektes, wonach bei Umstellung vom anaeroben auf aeroben Stoffwechsel bei gleichbleibendem Energiestatus der Zelle der Metabolitenstrom in der Glycolyse gedrosselt wird.
Regulatorfunktionen
Regulation in der Zelle
PFK1 besitzt ihr katalytisches Zentrum am N-Terminus, das regulatorische Zentrum am C-Terminus eines durch Genduplikation entstandenen Fusionsproteins. Beide Hälften zeigen infolgedessen Sequenzhomologien, unterlagen aber, entsprechend ihrer Aufgabe, getrennten Optimierungsprozessen:
- der katalytische Teil bindet die Substrate Fructose-6-phosphat (F-6-P) und ATP;
- ATP nimmt bei höheren Konzentrationen auch einen (niederaffinen) Bindungsplatz am regulatorischen Teil ein und wirkt von dort aus als allosterischer Inhibitor ("Enzymhemmung"). Eine Hemmfunktion teilt es mit weiteren endogenen Energieüberschusssignalen der Zelle (NADH/H+ und Citrat). Sind hingegen Energiemangelsignale (AMP, ADP) vorhanden, so wird das Enzym allosterisch aktiviert. Solange AMP und ADP vorherrschen, determinieren sie das Geschehen.
- In Erythrozyten wirkt das im Rapoport-Luebering-Zyklus durch das Enzym Bisphosphoglyceratmutase gebildete Intermediat 2,3-Diphosphoglycerat als ein Inhibitor der Phosphofructokinase.
Die Aktivität der Phosphofructokinase 1 in Muskelzellen wird auch durch den pH-Wert beeinflusst. Ein niedriger pH-Wert hemmt das Enzym und drosselt die Glykolyse. Dies passiert beispielsweise bei starker Muskelbeanspruchung, bei der viel Milchsäure entsteht. Diese senkt den pH-Wert in den Zellen.[10]
Regulation im Organismus
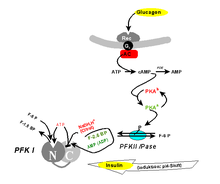
Seit längerem ist bekannt, dass PFK1 nicht nur durch eines seiner Substrate (ATP) inhibierbar ist, sondern auch durch eines seiner Produkte (F-1,6-BP) in vitro aktiviert werden kann („verkehrtes Enzym“). In der Zelle tritt der Letztere Effekt vermutlich nicht auf, da F-1,6-BP durch Aldolasetätigkeit nie die erforderliche Gleichgewichtskonzentration erreicht. Man fand jedoch, dass ein isomeres Molekül, das Fructose-2,6-bisphosphat (F-2,6-BP), ein physiologischer allosterischer Aktivator ist. F-2,6-BP vermittelt Hungersignale (zu niedriger Blutzucker), die vom Organismus über Glucagon oder Adrenalin ausgesandt werden. Nach Art eines "dritten Messengers" dient es zur Fortpflanzung entlang der Signaltransduktionskette Glucagon – cAMP – PKA (siehe „second messenger“).
F-2,6-BP ist das Produkt einer weiteren, spezialisierten Phosphofructokinase (PFKII). Diese „PFKII“, in Vertebraten ein Fusionsprotein aus Phosphofructokinase und Fructose-2,6-Bisphosphatase, gehört zu den interkonvertierbaren Enzymen, d.h. ihre Aktivität wird durch Proteinkinase A (PKA) und damit indirekt durch hormonelle Signale reguliert: Phosphorylierung eines einzigen Serinrestes schaltet die Kinaseaktivität ab, während gleichzeitig die Phosphataseaktivität angeschaltet wird. Das von Glucagon ausgehende Signal bewirkt also, dass F-2,6-BP nicht mehr verfügbar ist. Hierdurch kommt der Metabolitenstrom der Glykolyse an der PFKI zum Erliegen. In der Leber wird der resultierende G-6-P-Stau durch Überführung in Glucose abgebaut (bzw. die Glykolyse durch die Gluconeogenese umgekehrt), die als Neutralmolekül an den Blutkreislauf abgegeben werden kann. Das Glucagonsignal „zu geringer Blutzucker“ ist damit beantwortet.
Das gegenläufige (Insulin-)Signal "zu hoher Blutzucker" wird offenbar durch ein extrem pH-abhängiges Aktivitätsprofil realisiert. Als Antagonist des Glucagons hat das Insulin auch Wirkung auf die F-2,6-BP-Konzentration, indem über Aktivierung einer Phosphodiesterase der cAMP-Spiegel gesenkt und eine Phosphatase aktiviert wird. Diese dephosphoryliert die PFKII, sodass ihre Kinaseaktivität zum Tragen kommt und F-2,6-BP hergestellt wird, das aktivierend auf die PFKI und damit die Glykolyse wirkt. Damit wird die das Signal auslösende, überschüssige Blutglucose abgebaut. Dabei beinhaltet die Aktivierung der PFK1 nicht nur Konformationsänderungen der individuellen Untereinheiten, sondern auch Aggregatbildung zu höheren Oligomeren.
In Muskelzellen wirkt eine Phosphorylierung der PFKII nicht hemmend auf die Glykolyse, da hier Isoenzyme gebildet werden, deren Regulation in umgekehrter Richtung abläuft. Dies ist die Grundlage des Cori-Zyklus, über den bei Muskelaktivität unvollständig oxidiertes Lactat aus der Glykolyse über das Blut zur Leber gebracht wird, wo es (trotz gleicher hormoneller Situation) der Gluconeogenese zugeführt wird. In Muskelzellen hat außerdem, statt Glukagon, in erster Linie Adrenalin eine regulierende Funktion.[11]
Das Isoenzym im Skelettmuskel besitzt keine Phosphorylierungsstellen für die PKA, die eine Regulation über Phosphorylierung durch Hormone erlauben. Daher wirkt Adrenalin im Skelettmuskel nicht hemmend auf die Glykolyse und hemmt damit nicht die Glucoseverwertung, also die Energiegewinnung der Zellen.[11]
Im Herzmuskel hingegen findet wiederum eine Phosphorylierung statt. Diese bewirkt hier allerdings eine Stimulierung der Kinaseaktivität. Adrenalin bewirkt also eine Erhöhung der F-2,6-BP-Konzentration, und stimuliert damit die Glykolyse zusätzlich.[11]
Phosphofructokinase in der Photosynthese
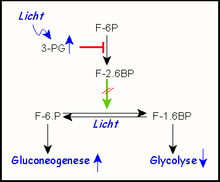
Bei der Photosynthese entsteht in Pflanzen durch Lichtenergie ATP und NADPH/H+ für Biosynthesen. Gleichzeitig entsteht durch Kohlendioxid-Fixierung (Assimilation) bei C3-Pflanzen 3-Phosphoglycerat (3-PG), ein Intermediat sowohl der Glycolyse als auch der Glucose-Biosynthese (Gluconeogenese). Bei Energieüberschuss ist der letztere Weg gefragt, der schließlich zum Energiespeicher Stärke führt. Verfügbarkeit von 3-PG reguliert (hemmt) PFKII, wodurch die Gluconeogenese ein- die Glycolyse aber ausgeschaltet wird.
- Energieüberschusssignale der Zelle (ATP, Citrat und NADH/H+ in tierischen, 3-PG in pflanzlichen Geweben) verhindern also allgemein die Bildung überflüssigen ATPs.
Klinische Bedeutung
Eine genetische Mutation im PFKM-Gen führt zur Tarui-Krankheit, einer Glykogenspeicherkrankheit, bei der die Fähigkeit bestimmter Zelltypen, Kohlenhydrate als Energiequelle zu nutzen, beeinträchtigt ist.[12]
Die Tarui-Krankheit ist eine Glykogenspeicherkrankheit mit Symptomen wie Muskelschwäche (Myopathie), bewegungsinduzierte Krämpfe und Spasmen, Myoglobinurie (Vorhandensein von Myoglobin im Urin, was auf Verletzungen der quergestreiften- und Herzmuskulatur hindeutet) und kompensierter Hämolyse. ATP ist ein natürlicher allosterischer Inhibitor von PFK, um eine unnötige Produktion von ATP durch Glykolyse zu verhindern. Eine Mutation in Asp(543)Ala kann jedoch zu einer stärkeren Hemmwirkung von ATP führen (aufgrund einer erhöhten Bindung an die hemmende allosterische Bindungsstelle von PFK).[13]
Damit Krebszellen aufgrund ihres schnellen Zellwachstums und ihrer schnellen Zellteilung ihren Energiebedarf decken können, überleben sie effektiver, wenn sie über eine hyperaktive Phosphofructokinase 1 verfügen. Wenn Krebszellen schnell wachsen und sich teilen, haben sie anfangs nicht viel Blut zur Verfügung und können daher eine Hypoxie (Sauerstoffmangel) aufweisen. Dies löst die O-GlcNAcylierung von Ser529 aus und gibt Krebszellen einen selektiven Wachstumsvorteil.[14][15]
Einige Viren, einschließlich HIV, HCMV und Mayaro beeinflussen zelluläre Stoffwechselwege wie die Glykolyse durch eine MOI-abhängige Erhöhung der Aktivität von PFK. Herpes-simplex-Viren (HSV) erhöhen die PFK-Aktivität durch Phosphorylierung des Enzyms an den Serinresten. Die HSV-1-induzierte Glykolyse erhöht den ATP-Gehalt, der für den Replikationsmechanismus des Virus entscheidend ist.[16]
Siehe auch
Einzelnachweise
- ↑ UniProt
 P08237
P08237
- ↑ Jeremy M. Berg, John L. Tymoczko, Lubert Stryer: Biochemistry.
6. Auflage. W. H. Freeman, 2007, ISBN 978-0-7167-8724-2 ( eingeschränkte Vorschau
in der Google-Buchsuche).
- ↑ G. A. Dunaway, T. P. Kasten, T. Sebo, R. Trapp: Analysis of the phosphofructokinase
subunits and isoenzymes in human tissues. In: The Biochemical journal, Band 251, Nummer 3, Mai 1988, S. 677–683,
doi:10.1042/bj2510677,
PMID 2970843,
PMC 1149058 (freier Volltext).
- ↑ P. R. Evans, G. W. Farrants, P. J. Hudson: Phosphofructokinase: structure and
control. In: Philosophical transactions of the Royal Society of London. Series B, Biological sciences, Band 293, Nummer 1063, Juni 1981, S. 53–62,
doi:10.1098/rstb.1981.0059,
PMID 6115424.
- ↑ Y. Shirakihara, P. R. Evans: Crystal structure of the complex of phosphofructokinase
from Escherichia coli with its reaction products. In: Journal of molecular biology, Band 204, Nummer 4, Dezember 1988, S. 973–994,
doi:10.1016/0022-2836(88)90056-3,
PMID 2975709.
- ↑ K. Banaszak, I. Mechin, G. Obmolova, M. Oldham, S. H. Chang, T. Ruiz, M. Radermacher,
G. Kopperschläger, W. Rypniewski: The crystal structures of eukaryotic phosphofructokinases from baker’s yeast and rabbit skeletal muscle. In:
Journal of molecular biology. Band 407, Nummer 2, März 2011, S. 284–297,
doi:10.1016/j.jmb.2011.01.019,
PMID 21241708.
- ↑ Sophie T. Williams, Alex Gutteridge, Craig Porter, Gemma L. Holliday, Morwenna Hall:
Phosphofructokinase I. In: Mechanism and Catalytic Site Atlas.
EMBL-EBI,
- ↑ K. Peskov, I. Goryanin, O. Demin: Kinetic model of phosphofructokinase-1
from Escherichia coli. In: Journal of bioinformatics and computational biology. Band 6, Nummer 4, August 2008, S. 843–867,
PMID 18763746.
- ↑ H. W. Hellinga, P. R. Evans: Mutations in the active site of Escherichia coli
phosphofructokinase. In: Nature, Band 327, Nummer 6121, 1987 Jun 4-10, S. 437–439,
doi:10.1038/327437a0,
PMID 2953977.
- ↑ H. Robert Horton, Laurence A. Moran, K. Gray Scrimgeour, Marc D. Perry, J. David Rawn, Carsten Biele (Übersetzer): Biochemie. Pearson Studium. 4. aktualisierte Auflage. 2008, ISBN 978-3-8273-7312-0, S. 468.
- ↑ Hochspringen nach: a b c Joachim Rassow, Karin Hauser, Roland Netzker, Rainer Deutzmann: Duale Reihe: Biochemie. 2. Auflage. Thieme Verlag, Stuttgart 2008, ISBN 978-3-13-125352-1, S. 219 f.
- ↑ H. Nakajima, N. Raben, T. Hamaguchi, T. Yamasaki: Phosphofructokinase deficiency;
past, present and future. In: Current molecular medicine, Band 2, Nummer 2, März 2002, S. 197–212,
doi:10.2174/1566524024605734,
PMID 11949936 (Review).
- ↑ A. Brüser, J. Kirchberger, T. Schöneberg: Altered allosteric regulation of muscle
6-phosphofructokinase causes Tarui disease. In: Biochemical and biophysical research communications, Band 427, Nummer 1, Oktober 2012, S. 133–137,
doi:10.1016/j.bbrc.2012.09.024,
PMID 22995305.
- ↑ W. Yi, P. M. Clark, D. E. Mason, M. C. Keenan, C. Hill, W. A. Goddard, E. C. Peters,
E. M. Driggers, L. C. Hsieh-Wilson: Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. In:
Science, Band 337, Nummer 6097, August 2012, S. 975–980,
doi:10.1126/science.1222278,
PMID 22923583,
PMC 3534962 (freier Volltext).
- ↑ L. S. Gomez, P. Zancan, M. C. Marcondes, L. Ramos-Santos, J. R. Meyer-Fernandes,
M. Sola-Penna, D. Da Silva: Resveratrol decreases breast cancer cell viability and glucose metabolism by inhibiting 6-phosphofructo-1-kinase. In: Biochimie,
Band 95, Nummer 6, Juni 2013, S. 1336–1343,
doi:10.1016/j.biochi.2013.02.013,
PMID 23454376.
- ↑ J. L. Abrantes, C. M. Alves, J. Costa, F. C. Almeida, M. Sola-Penna, C. F. Fontes,
T. M. Souza: Herpes simplex type 1 activates glycolysis through engagement of the enzyme 6-phosphofructo-1-kinase (PFK-1). In:
Biochimica et Biophysica Acta, Band 1822, Nummer 8, August 2012, S. 1198–1206;
doi:10.1016/j.bbadis.2012.04.011,
PMID 22542512.


© biancahoegel.de
Datum der letzten Änderung: Jena, den: 19.06. 2026