Massenwirkungsgesetz
Das Massenwirkungsgesetz (Abkürzung „MWG“) definiert das chemische
Gleichgewicht für chemische
Reaktionen. Dem Massenwirkungsgesetz zufolge ist das Produkt aus den Aktivitäten
 der beteiligten Stoffe
der beteiligten Stoffe  (potenziert mit den jeweiligen stöchiometrischen
Zahlen
(potenziert mit den jeweiligen stöchiometrischen
Zahlen  )
konstant.
Die Konstante wird als Gleichgewichts- oder
Massenwirkungskonstante
)
konstant.
Die Konstante wird als Gleichgewichts- oder
Massenwirkungskonstante  bezeichnet.
Sind an einer Reaktion
bezeichnet.
Sind an einer Reaktion  Stoffe beteiligt, erhält man:
Stoffe beteiligt, erhält man:

Hierbei ist  das Produktzeichen.
Da die stöchiometrischen Zahlen der Ausgangsstoffe negativ sind, werden
Massenwirkungsgesetze als Brüche
formuliert, wobei die Produktterme den Zähler und die Eduktterme den Nenner
bilden. Für eine Reaktion mit der unter Verwendung des Gleichgewichtspfeils
formulierten stöchiometrischen Reaktionsgleichung
das Produktzeichen.
Da die stöchiometrischen Zahlen der Ausgangsstoffe negativ sind, werden
Massenwirkungsgesetze als Brüche
formuliert, wobei die Produktterme den Zähler und die Eduktterme den Nenner
bilden. Für eine Reaktion mit der unter Verwendung des Gleichgewichtspfeils
formulierten stöchiometrischen Reaktionsgleichung
 ,
,
an der die Ausgangsstoffe A, B … M und die Produkte N, O … Z beteiligt sind, nimmt das Massenwirkungsgesetz daher folgende Form an:

Die Gleichgewichtskonstante ist eine intensive dimensionslose thermodynamische Zustandsgröße und definiert diejenige stoffliche Zusammensetzung, für die das unter den jeweiligen Reaktionsbedingungen relevante thermodynamische Potential des reagierenden Systems ein Minimum aufweist. Stellt sich der durch das Massenwirkungsgesetz definierte Gleichgewichtszustand ein, wird die maximale Zunahme der Entropie, die durch Zustandsänderungen im Reaktionsgemisch realisierbar ist, erreicht.
Geschichte

Das Massenwirkungsgesetz wurde von den norwegischen Chemikern Cato Maximilian Guldberg und Peter Waage experimentell ermittelt und 1864 in Norwegisch sowie 1867 in Französisch (mit ihren experimentellen Daten und einem modifizierten Gesetz) erstmals veröffentlicht. Ihre Veröffentlichung fand lange keine große Beachtung. Der Ire John Hewitt Jellett kam 1873 zu ähnlichen Schlussfolgerungen, ebenso wie 1877 Jacobus Henricus van ’t Hoff. Insbesondere nach den Veröffentlichungen von van ’t Hoff (aber auch von anderen wie August Friedrich Horstmann) hatten Guldberg und Waage den Eindruck, dass ihre Arbeit nicht genug bekannt sei. Nachdem sie 1879 im Journal für Praktische Chemie eine ausführlichere Darlegung in deutscher Sprache veröffentlicht hatten, erkannte van ’t Hoff deren Priorität an.
Thermodynamische Aspekte
Thermodynamische Definition der Gleichgewichtskonstante
Im thermodynamischen Gleichgewicht ist die Änderung des (zur Beschreibung des Systems angebrachten) thermodynamischen Potentials null. Die Gleichgewichtskonstante hängt dann lediglich von den gewählten (willkürlichen, aber festen) Referenzzuständen (o) der beteiligten Stoffe ab. Der Referenzzustand (o) kann entsprechend dem betrachteten Reaktionsszenario frei gewählt werden und ist nicht mit sogenannten Standardzuständen unter Standardbedingungen zu verwechseln. Die Lage des Gleichgewichtes sowie der Wert der Gleichgewichtskonstante hängen dabei vom gewählten Referenzzustand ab.
Da thermodynamische Gleichgewichtszustände unabhängig vom Weg sind, auf dem diese erreicht werden, ist es für die thermodynamische Gültigkeit des Massenwirkungsgesetzes nicht erforderlich, die unabhängigen Zustandsvariablen des relevanten thermodynamischen Potentials während des gesamten Reaktionsverlaufes konstant zu halten.
In der Praxis sind vor allem zwei Szenarien von Bedeutung:
Reaktionen, die in Autoklaven
durchgeführt werden, wie etwa Solvothermalsynthesen,
finden bei konstantem Volumen
und in der Regel bei konstanter Temperatur
statt, wohingegen der Druck
variabel ist. Wird eine Reaktion bei konstanter Temperatur, konstantem Volumen
und variablem Druck durchgeführt, ist die freie
Energie (Helmholtz-Potential) F das relevante thermodynamische
Potential, da neben der sich im Verlauf der chemischen Reaktion verändernden
stofflichen Zusammensetzung des reagierenden Systems Temperatur und Volumen die
unabhängigen Zustandsvariablen der freien Energie sind. Das Massenwirkungsgesetz
definiert dann die stoffliche Zusammensetzung des reagierenden Systems, für die
die freie Energie minimal wird und die damit den thermodynamischen
Gleichgewichtszustand darstellt. Für eine chemische Reaktion, die in einem durch
ein konstantes Volumen und eine konstante Temperatur gekennzeichneten
Referenzzustand (o) durchgeführt wird und deren
beteiligte Komponenten
in diesem Referenzzustand die molaren
freien Bildungsenergien  besitzen, wird die molare freie Reaktionsenergie
besitzen, wird die molare freie Reaktionsenergie  gleich:
gleich:

Die Gleichgewichtskonstante
ist dann unter Verwendung der absoluten
Temperatur  wie folgt durch die molare freie Reaktionsenergie des Referenzzustandes
definiert:
wie folgt durch die molare freie Reaktionsenergie des Referenzzustandes
definiert:
![{\displaystyle K=\exp {\bigg [}-{\frac {\Delta _{R}F^{\circ }}{R\cdot T}}{\bigg ]}.}](/svg/4f3e424aed7a605a22a69601cff19cfdb3ba95b7.svg)
Viele chemische Reaktionen werden in offenen Systemen durchgeführt, so dass
Druckausgleich zwischen dem reagierenden System und der Umgebung erfolgen kann.
Somit kann angenommen werden, dass die betrachtete Reaktion unter einem
konstanten, dem Umgebungsdruck entsprechenden Druck durchgeführt wird, während
das Volumen des reagierenden Systems variabel ist. Wird eine Reaktion bei
konstantem Druck, konstanter Temperatur und variablem Volumen durchgeführt, ist
die freie Enthalpie
(Gibbs-Energie) das relevante thermodynamische Potential, da neben der sich
im Verlauf der chemischen Reaktion verändernden stofflichen Zusammensetzung des
reagierenden Systems Druck und Temperatur die unabhängigen Zustandsvariablen der
freien Enthalpie sind. In diesem Fall definiert das Massenwirkungsgesetz die
stoffliche Zusammensetzung des reagierenden Systems, für die die freie Enthalpie
minimal wird und die damit den thermodynamischen Gleichgewichtszustand
darstellt. Für eine chemische Reaktion, die in einem durch einen konstanten
Druck und eine konstante Temperatur gekennzeichneten Referenzzustand (o)
durchgeführt wird und deren
beteiligte Komponenten
in diesem Referenzzustand die molaren freien Bildungsenthalpien  besitzen, wird die molare freie Reaktionsenthalpie
besitzen, wird die molare freie Reaktionsenthalpie  gleich:
gleich:

Die Gleichgewichtskonstante
ist dann wie folgt durch die molare freie Reaktionsenthalpie der
Referenzzustände
definiert:
![{\displaystyle K=\exp {\bigg [}-{\frac {\Delta _{R}G^{\circ }}{R\cdot T}}{\bigg ]}}](/svg/c427cdc476feaef020c6054e2041215d36f15625.svg)
Wird ein chemisches Gleichgewicht durch die Änderung der Konzentration eines an der betrachteten Reaktion beteiligten Stoffes – und damit seiner Aktivität – gestört, müssen sich gemäß dem Prinzip vom kleinsten Zwang nach Henry Le Chatelier die Aktivitäten (und damit die Konzentrationen) der anderen an der Reaktion beteiligten Stoffe so ändern, dass das Massenwirkungsgesetz erfüllt bleibt. Die Gleichgewichtskonstante ist somit unabhängig von den Ausgangskonzentrationen der an der betrachteten Reaktion beteiligten Stoffe. Als thermodynamische Zustandsgröße hängt die Gleichgewichtskonstante nicht vom Reaktionsmechanismus oder von kinetischen Größen wie Geschwindigkeitskonstanten und Reaktionsgeschwindigkeiten ab.
Herleitung
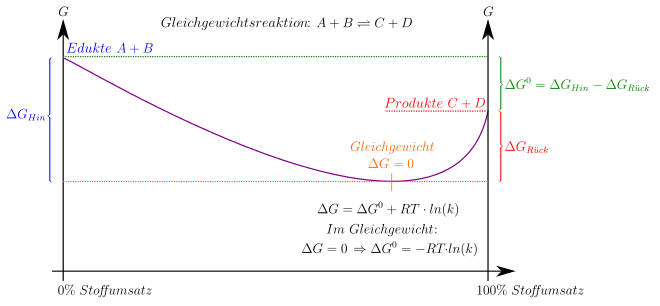
Im Folgenden
wird exemplarisch angenommen, dass die betrachtete Reaktion bei einem durch
einen konstanten Druck sowie eine konstante Temperatur gekennzeichneten
Referenzzustand ( )
durchgeführt wird, so dass die freie Enthalpie das relevante thermodynamische
Potential ist. Sinngemäß lässt sich die unten skizzierte Herleitung auf jedes
andere thermodynamische Potential anwenden.
)
durchgeführt wird, so dass die freie Enthalpie das relevante thermodynamische
Potential ist. Sinngemäß lässt sich die unten skizzierte Herleitung auf jedes
andere thermodynamische Potential anwenden.
 sei der Beitrag eines Stoffes
sei der Beitrag eines Stoffes  zur extensiven freien Gesamtenthalpie
zur extensiven freien Gesamtenthalpie  des betrachteten Systems. Sofern Druck, Temperatur und die Stoffmengen
des betrachteten Systems. Sofern Druck, Temperatur und die Stoffmengen  aller weiteren anwesenden Stoffe
aller weiteren anwesenden Stoffe  konstant gehalten werden, ist das chemische
Potential
konstant gehalten werden, ist das chemische
Potential  des Stoffes
gleich der Änderung
des Stoffes
gleich der Änderung  pro Änderung
pro Änderung  der Stoffmenge
der Stoffmenge  des Stoffes :
des Stoffes :

Die Menge der Formelumsätze einer chemischen Reaktion in Mol
ist durch die Umsatzvariable
 gegeben, dem Quotienten aus
und der stöchiometrischen
Zahl
des Stoffes :
gegeben, dem Quotienten aus
und der stöchiometrischen
Zahl
des Stoffes :

Ersetzt man im Ausdruck für das chemische Potential
durch  und löst nach
auf, erhält man:
und löst nach
auf, erhält man:

Enthält ein Reaktionsgemisch insgesamt
verschiedene Stoffe, gilt für die Änderung  der freien Gesamtenthalpie
des reagierenden Systems:
der freien Gesamtenthalpie
des reagierenden Systems:

Dividieren durch  ergibt:
ergibt:

Unter den gegebenen Reaktionsbedingungen für  ist
ist  die Änderung der freien Systementhalpie
pro Reaktionsumsatz.
die Änderung der freien Systementhalpie
pro Reaktionsumsatz.
Das chemische Potential
eines Stoffes
in einer Reaktionsmischung kann bezogen auf das chemische Potential  des reinen Stoffes
für den Referenzzustand ()
ausgedrückt werden, der für das betrachtete Reaktionsszenario maßgeblich
ist:
des reinen Stoffes
für den Referenzzustand ()
ausgedrückt werden, der für das betrachtete Reaktionsszenario maßgeblich
ist:

Hierbei ist  die relative chemische
Aktivität mit der Gaskonstante
die relative chemische
Aktivität mit der Gaskonstante
 und der absoluten Temperatur .
und der absoluten Temperatur .
Die Kombination mit obigem Ausdruck für  ergibt:
ergibt:

Die Summe  ist gleich der molaren freien Reaktionsenthalpie
für den Referenzzustand. Man erhält:
ist gleich der molaren freien Reaktionsenthalpie
für den Referenzzustand. Man erhält:

Während
konstant ist, solange Druck und Temperatur nicht verändert werden, hängt die
Summe  von den jeweils aktuellen relativen Aktivitäten
ab. Durch Anwendung der einschlägigen Logarithmusregel
lässt sich die Summe
in den Logarithmus des Produkts
von den jeweils aktuellen relativen Aktivitäten
ab. Durch Anwendung der einschlägigen Logarithmusregel
lässt sich die Summe
in den Logarithmus des Produkts  umwandeln, welches als Reaktionsquotient
umwandeln, welches als Reaktionsquotient
 bezeichnet wird:
bezeichnet wird:

Somit ergibt sich:

Solange im Verlauf der betrachteten Reaktion die transienten relativen
Aktivitäten der Edukte größer als die relativen Gleichgewichtsaktivitäten der
Edukte sind, ist
kleiner als die Gleichgewichtskonstante ,
und es gilt:

Folglich nimmt
mit Fortlaufen der Reaktion ab:

Im chemischen Gleichgewicht nimmt die freie Gesamtenthalpie
des reagierenden Systems den kleinstmöglichen erreichbaren Wert an. Im
chemischen Gleichgewicht weist die Funktion  somit ein Minimum auf. Der partielle Differentialquotient
somit ein Minimum auf. Der partielle Differentialquotient  muss demnach im chemischen Gleichgewicht gleich null sein:
muss demnach im chemischen Gleichgewicht gleich null sein:

Der Reaktionsquotient
ist im chemischen Gleichgewicht allein durch die freie Reaktionsenthalpie im
Referenzzustand
gegeben und entspricht damit der Gleichgewichtskonstanten :

Dieser Ausdruck stellt den Zusammenhang zwischen der molaren freien
Reaktionsenthalpie
und der stofflichen Zusammensetzung des Reaktionsgemisches im chemischen
Gleichgewicht dar. Auflösen nach
ergibt entsprechend:
![{\displaystyle K=\exp {\bigg [}-{\frac {\Delta _{R}G^{\circ }}{R\cdot T}}{\bigg ]}=\prod _{i=1}^{z}a_{i}^{{\nu }_{i}}.}](/svg/53d49263b7305bae254b155e8aa0381fcdd534b5.svg)
Da
von
abhängt, hängt auch der Zahlenwert der Gleichgewichtskonstante vom jeweils
angewendeten Referenzzustand ()
ab.
Druck- und Temperaturabhängigkeit der Gleichgewichtskonstante
Da sich das Massenwirkungsgesetz auf ein unter den jeweiligen
Reaktionsbedingungen anzuwendendes thermodynamisches Potential bezieht, ist die
Gleichgewichtskonstante abhängig von den Zustandsgrößen, die die unabhängigen
Zustandsvariablen des betreffenden thermodynamischen Potentials darstellen. Ist
das relevante thermodynamische Potential die freie Enthalpie, ändert sich der
Wert der Gleichgewichtskonstante, wenn die betrachtete Reaktion bei
unverändertem Druck und einer veränderten konstanten Temperatur durchgeführt
wird. Die Temperaturabhängigkeit der Gleichgewichtskonstante bei konstantem
Druck lässt sich durch die van-’t-Hoff-Gleichung
beschreiben beziehungsweise durch van-'t-Hoffsche Reaktionsisobaren darstellen.
Ebenso ändert sich der Wert der Gleichgewichtskonstanten, wenn die betrachtete
Reaktion bei unveränderter Temperatur und verändertem konstanten Druck
durchgeführt wird. Zur Beschreibung der Druckabhängigkeit der
Gleichgewichtskonstante
bei konstanter Temperatur in kondensierten Phasen wird das molare
Reaktonsvolumen  herangezogen:
herangezogen:

Die Druckabhängigkeit der Gleichgewichtskonstante ist bei Reaktionen, die in kondensierten Phasen stattfinden, jedoch typischerweise sehr schwach und wird häufig vernachlässigt. Ist das relevante thermodynamische Potential die freie Energie, wird die Temperaturabhängigkeit der Gleichgewichtskonstante bei konstantem Volumen durch die van-’t-Hoff’sche Reaktionsisochore beschrieben.
Massenwirkungsgesetz als Spezialfall in der Reaktionskinetik
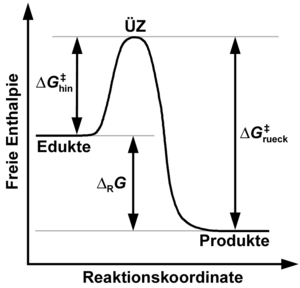
 aufgebracht werden. Durchläuft die Rückreaktion denselben Übergangszustand wie
die Hinreaktion, ist die freie Aktivierungsenthalpie der Rückreaktion
aufgebracht werden. Durchläuft die Rückreaktion denselben Übergangszustand wie
die Hinreaktion, ist die freie Aktivierungsenthalpie der Rückreaktion  gleich
gleich  (freie Reaktionsenthalpie).
(freie Reaktionsenthalpie).Gemäß der Theorie
des Übergangszustandes müssen im Verlauf elementarer Reaktionsereignisse
Ausgangsstoffe und Produkte trennende Potentialbarrieren überwunden werden, die
sich auf der makroskopischen Ebene am zweckmäßigsten durch das jeweils
anzuwendende thermodynamische Potential beschreiben lassen. Reversible
Reaktionen zeichnen sich dadurch aus, dass neben Hinreaktionen, die zur
Bildung der Reaktionsprodukte aus den Ausgangsstoffen führen, auch
Rückreaktionen, die zur Bildung der Ausgangsstoffe aus den Reaktionsprodukten
führen, in nennenswertem Umfang stattfinden. Werden beispielsweise Druck und
Temperatur konstant gehalten, repräsentiert die molare freie
Aktivierungsenthalpie  die Höhe der bei der Hinreaktion für die Umwandlung der Ausgangsstoffe in die
Produkte zu überwindenden Potentialbarriere (bezogen auf den Referenzzustand).
Entsprechend repräsentiert die molare freie Aktivierungsenthalpie
die Höhe der bei der Hinreaktion für die Umwandlung der Ausgangsstoffe in die
Produkte zu überwindenden Potentialbarriere (bezogen auf den Referenzzustand).
Entsprechend repräsentiert die molare freie Aktivierungsenthalpie  die Höhe der bei der Rückreaktion für die Umwandlung der Produkte in die
Ausgangsstoffe zu überwindenden Potentialbarriere. Hin- und Rückreaktion können,
müssen aber nicht entlang derselben
Reaktionstrajektorie
verlaufen. Sofern Hin- und Rückreaktion exakt entlang derselben
Reaktionstrajektorie in jeweils entgegengesetzter Richtung ablaufen, gilt mit
die Höhe der bei der Rückreaktion für die Umwandlung der Produkte in die
Ausgangsstoffe zu überwindenden Potentialbarriere. Hin- und Rückreaktion können,
müssen aber nicht entlang derselben
Reaktionstrajektorie
verlaufen. Sofern Hin- und Rückreaktion exakt entlang derselben
Reaktionstrajektorie in jeweils entgegengesetzter Richtung ablaufen, gilt mit
 als molarer freier Reaktionsenthalpie der Hinreaktion für :
als molarer freier Reaktionsenthalpie der Hinreaktion für :

Die Kinetik einer betrachteten Reaktion wird durch ein Geschwindigkeitsgesetz
beschrieben, in das das thermodynamische Aktivierungspotential mittels einer
Geschwindigkeitskonststante eingeht. Die Geschwindigkeitskonstante der
Hinreaktion  ,
die die Kinetik der Umwandlung der Ausgangsstoffe in die Produkte repräsentiert,
hängt von
wie folgt ab (siehe Abschnitt "Thermodynamische
Formulierung" im Artikel "Theorie des Übergangszustandes")-->:
,
die die Kinetik der Umwandlung der Ausgangsstoffe in die Produkte repräsentiert,
hängt von
wie folgt ab (siehe Abschnitt "Thermodynamische
Formulierung" im Artikel "Theorie des Übergangszustandes")-->:
![{\displaystyle k_{\mathrm {hin} }=\mathrm {Konstante} \cdot \exp {\bigg [}-{\frac {\Delta {G^{\ddagger }}_{\mathrm {hin} }^{\circ }}{RT}}{\Bigg ]}}](/svg/0c7fb33baa2d5a07d4ed9076f0df8d8a6f9217d7.svg)
Entsprechend gilt für die Geschwindigkeitskonstante  ,
die die Kinetik der Umwandlung der Produkte die Ausgangsstoffe durch die
Rückreaktion repräsentiert:
,
die die Kinetik der Umwandlung der Produkte die Ausgangsstoffe durch die
Rückreaktion repräsentiert:
![{\displaystyle k_{\text{rück}}=\mathrm {Konstante} \cdot \exp {\bigg [}-{\frac {\Delta {G^{\ddagger }}_{\text{rück}}^{\circ }}{RT}}{\Bigg ]}}](/svg/8a8ad8733aca870284669d7ee4aa68ada38b9595.svg)
Durch Anwendung des Ausdrucks  lässt sich
als Funktion von
und
darstellen:
lässt sich
als Funktion von
und
darstellen:
![{\displaystyle k_{\text{rück}}=\mathrm {Konstante} \cdot \exp {\Bigg [}-{\frac {\Delta {G^{\ddagger }}_{\text{rück}}^{\circ }}{RT}}{\Bigg ]}={\rm {Konstante}}\cdot \exp {\Bigg [}-{\frac {\Delta {G^{\ddagger }}_{\mathrm {hin} }^{\circ }-\Delta _{\mathrm {R} }G^{\circ }}{RT}}{\Bigg ]}}](/svg/959e3f4255a3213f75c35d84fc3bdb7957f81847.svg)
Für den Quotienten aus
und
folgt:
![{\displaystyle {\frac {k_{\mathrm {hin} }}{k_{\text{rück}}}}=\exp {\Bigg [}-{\frac {\Delta {G^{\ddagger }}_{\mathrm {hin} }^{\circ }}{RT}}{\Bigg ]}\cdot \exp {\Bigg [}-{\frac {\Delta {G^{\ddagger }}_{\mathrm {hin} }^{\circ }-\Delta _{\mathrm {R} }G^{\circ }}{RT}}{\Bigg ]}^{-1}=\exp {\Bigg [}-{\frac {\Delta _{\mathrm {R} }G^{\circ }}{RT}}{\Bigg ]}=K}](/svg/18223a9bc291a4311cc7bc6472789b7f1cb8bdbb.svg)
Das Verhältnis  ist somit gleich der Gleichgewichtskonstante
ist somit gleich der Gleichgewichtskonstante  und wird durch die Referenz freie Reaktionsenthalpie
vorgegeben. Der Zusammenhang zwischen der Gleichgewichtskonstante und den
Geschwindigkeitskonstanten der Hin- und Rückreaktion ist thermodynamisch
begründet und gilt unabhängig von der Art und Weise, in der die die
Reaktionskinetik beschreibenden Geschwindigkeitsgesetze formuliert werden.
und wird durch die Referenz freie Reaktionsenthalpie
vorgegeben. Der Zusammenhang zwischen der Gleichgewichtskonstante und den
Geschwindigkeitskonstanten der Hin- und Rückreaktion ist thermodynamisch
begründet und gilt unabhängig von der Art und Weise, in der die die
Reaktionskinetik beschreibenden Geschwindigkeitsgesetze formuliert werden.
Entsprechen für eine reversible Reaktion

lassen sich mit  sowie
sowie  als Reaktionsgeschwindigkeiten der Hin- und Rückreaktion folgende
Geschwindigkeitsgesetze formulieren (siehe auch Ratengleichung):
als Reaktionsgeschwindigkeiten der Hin- und Rückreaktion folgende
Geschwindigkeitsgesetze formulieren (siehe auch Ratengleichung):


Teilt man den Ausdruck für
durch den Ausdruck für ,
erhält man:

Mit  sowie
sowie  erhält man:
erhält man:

Daraus folgt, dass im chemischen Gleichgewicht die Geschwindigkeit der
Hinreaktion
gleich der Geschwindigkeit der Rückreaktion
sein muss:

Aufstellung des Massenwirkungsgesetzes mit Stoffmengenkonzentrationen
Die relativen
Aktivitäten  sind relativ zum Referenzzustand (mit Referenzkonzentration
sind relativ zum Referenzzustand (mit Referenzkonzentration  ),
wobei der Aktivitätskoeffizient
),
wobei der Aktivitätskoeffizient
 für wechselwirkende Systeme ungleich eins ist. Durch Einsetzen der relativen
Aktivitäten erhält man
für wechselwirkende Systeme ungleich eins ist. Durch Einsetzen der relativen
Aktivitäten erhält man

Unter Vernachlässigung der Teilchenwechselwirkungen – d.h. durch
Fordern, dass die Aktivitätskoeffizienten  – erhält man
– erhält man

was für verdünnte Lösungen häufig eine gute Näherung ist. Teilweise werden die Referenzkonzentrationen zusätzlich in die Definition einer neuen Gleichgewichtskonstante einbezogen:

Statt thermodynamisch korrekt mit den Aktivitäten der an der betrachteten Reaktion beteiligten Stoffe kann somit das Massenwirkungsgesetz für Reaktionen in verdünnter Lösung häufig näherungsweise unter Verwendung der Stoffmengenkonzentrationen der beteiligten Stoffe aufgestellt werden. Bei stärker konzentrierten Lösungen können jedoch die Werte der Aktivitätskoeffizienten stark von 1 abweichen, so dass diese Näherung mit Vorsicht zu gebrauchen ist. Die mit Stoffmengenkonzentrationen berechneten Gleichgewichtskonstanten werden zur Unterscheidung von mit Aktivitäten berechneten Gleichgewichtskonstanten mit Kc bezeichnet, wobei der tiefgestellte Index c für Stoffmengenkonzentration steht. Das Massenwirkungsgesetz wird zum Beispiel für die Reaktion
unter Verwendung der Stoffmengenkonzentrationen c(A), c(B), c(C) und c(D) der Ausgangsstoffe A und B sowie der Produkte C und D wie folgt formuliert:

Da die stöchiometrischen Zahlen  und
und  der Ausgangsstoffe A und B als Exponenten von deren Stoffmengenkonzentrationen
c(A) und c(B) definitionsgemäß ein negatives Vorzeichen besitzen,
stehen die Produktterme
der Ausgangsstoffe A und B als Exponenten von deren Stoffmengenkonzentrationen
c(A) und c(B) definitionsgemäß ein negatives Vorzeichen besitzen,
stehen die Produktterme  und
und  im
Nenner des Ausdrucks für
im
Nenner des Ausdrucks für  .
.
Eine mittels der Stoffmengenkonzentrationen erhaltene Gleichgewichtskonstante
hat in der Regel einen anderen Zahlenwert als die entsprechende mittels der
Aktivitäten erhaltene Gleichgewichtskonstante. Weiterhin kann
eine Dimension
und damit auch eine Einheit
besitzen.  sei die Summe aller stöchiometrischer Zahlen
der beteiligten Stoffe
einer Reaktion mit
sei die Summe aller stöchiometrischer Zahlen
der beteiligten Stoffe
einer Reaktion mit  beteiligten Stoffen:
beteiligten Stoffen:

 ist das Dimensionssymbol für Länge,
ist das Dimensionssymbol für Länge,
 das Dimensionssymbol für die Stoffmenge. Die Dimension der
Stoffmengenkonzentration ist
das Dimensionssymbol für die Stoffmenge. Die Dimension der
Stoffmengenkonzentration ist  und die Dimension von
demzufolge
und die Dimension von
demzufolge  .
Lediglich wenn
.
Lediglich wenn  ist, ist
dimensionslos. Betrachtet man beispielsweise die Synthese von
Kaliumhexacyanidoferrat(II)
gemäß
ist, ist
dimensionslos. Betrachtet man beispielsweise die Synthese von
Kaliumhexacyanidoferrat(II)
gemäß
![{\displaystyle \mathrm {Fe^{2+}+6\ CN^{-}\rightleftharpoons [Fe(CN)_{6}]^{4-}} }](/svg/b7776968d1e09aa0c3196c43f8d99c4c94bdcdf9.svg) ,
,
ergibt sich für :
![{\displaystyle K_{c}=\mathrm {\frac {c([Fe(CN)_{6}]^{4-})}{c(Fe^{2+})\cdot c^{6}(CN^{-})}} }](/svg/1964a7d5b0996e48175befe55b4b68c03d6c3cfa.svg)
In diesem Beispiel ist  und die Dimension von
gleich
und die Dimension von
gleich  .
Die Einheit von
ist demnach
.
Die Einheit von
ist demnach  .
.
Um eine dimensionslose Form der stoffmengenkonzentrationsbasierten
Gleichgewichtskonstante
zu erhalten, kann
durch die Einheitskonzentration  potenziert mit
dividiert werden.
potenziert mit
dividiert werden.
Aufstellung des Massenwirkungsgesetzes für homogene Gasgleichgewichte
Für Gasphasenreaktionen wird das Massenwirkungsgesetz häufig mit den Partialdrücken  der beteiligten Stoffe
aufgestellt. Als Symbol für mit Partialdrücken erhaltene
Gleichgewichtskonstanten wird
der beteiligten Stoffe
aufgestellt. Als Symbol für mit Partialdrücken erhaltene
Gleichgewichtskonstanten wird  verwendet. Bei homogenen Gasgleichgewichten mit
beteiligten Komponenten nimmt das Massenwirkungsgesetz entsprechend folgende
Form an:
verwendet. Bei homogenen Gasgleichgewichten mit
beteiligten Komponenten nimmt das Massenwirkungsgesetz entsprechend folgende
Form an:

Bei der Bildung von Iodwasserstoff aus elementarem Wasserstoff und Iod

stellt sich das Gleichgewicht

ein.
und  beziehungsweise die Partialdrücke
und die Stoffmengenkonzentrationen
beziehungsweise die Partialdrücke
und die Stoffmengenkonzentrationen  lassen sich über die Zustandsgleichung
für ideale Gase miteinander verknüpfen:
lassen sich über die Zustandsgleichung
für ideale Gase miteinander verknüpfen:


Für die Gleichgewichtskonstante
bei der Bildung von Iodwasserstoff ergibt sich:
![{\displaystyle K_{p}={\frac {[c\mathrm {(HI)} RT]^{2}}{c\mathrm {(H_{2})} RT\cdot \ c\mathrm {(I_{2})} RT}}={\frac {c^{2}\mathrm {(HI)} }{c\mathrm {(H_{2})} \cdot c\mathrm {(I_{2})} }}=K_{c}}](/svg/94a653e9a44e1bb3f34ec58261a8543c35e15257.svg)
Ist in einem Gasphasengleichgewicht die Teilchenanzahl der Produkte gleich
der Teilchenanzahl der Edukte, so kürzt sich  im mit Stoffmengenkonzentrationen formulierten Massenwirkungsgesetz heraus.
Betrachtet man jedoch die Reaktion von Schwefeldioxid
und Sauerstoff zu
Schwefeltrioxid
im mit Stoffmengenkonzentrationen formulierten Massenwirkungsgesetz heraus.
Betrachtet man jedoch die Reaktion von Schwefeldioxid
und Sauerstoff zu
Schwefeltrioxid
 mit
mit 
und ersetzt die Drücke durch Stoffmengenkonzentrationen, ergibt sich:
![K_{p}={\frac {[c\mathrm {(SO_{3})} RT]^{2}}{[c\mathrm {(SO_{2})} RT]^{2}\cdot c\mathrm {(O_{2})} RT}}={\frac {c^{2}\mathrm {\mathrm {(SO_{3})} } }{c^{2}\mathrm {(SO_{2})} \cdot c\mathrm {(O_{2})} }}\cdot {\frac {1}{RT}}=K_{c}\cdot {\frac {1}{RT}}](/svg/a46042c9ec04f3b2bcb98d9ba1a32affa1b9cc03.svg)
Die Teilchenzahl vermindert sich bei der Reaktion und ein Faktor  verbleibt im mit Stoffmengenkonzentrationen formulierten Massenwirkungsgesetz.
verbleibt im mit Stoffmengenkonzentrationen formulierten Massenwirkungsgesetz.
Betrachtet man die Bildung von Ammoniak im Haber-Bosch-Verfahren gemäß
 ,
,
ergibt sich:

Die Umrechnung von  in
erfolgt gemäß:
in
erfolgt gemäß:
![K_{p}={\frac {[c\mathrm {(NH_{3})} RT]^{2}}{c\mathrm {(N_{2})} RT\cdot [c\mathrm {(H_{2})} RT]^{3}}}={\frac {c^{2}\mathrm {(NH_{3})} }{c\mathrm {(N_{2})} \cdot c^{3}\mathrm {(H_{2})} }}\cdot {\frac {1}{(RT)^{2}}}=K_{c}\cdot {\frac {1}{(RT)^{2}}}](/svg/409569cdd708a93b2970f0fb1590194b200ed1bc.svg)
Allgemein lässt sich also das Massenwirkungsgesetz eines Gasphasengleichgewichts ausdrücken als:

Dabei ist  die Summe der stöchiometrische Zahlen der betrachteten Reaktion. Im Fall der
Bildung von HI aus den Elementen ist
die Summe der stöchiometrische Zahlen der betrachteten Reaktion. Im Fall der
Bildung von HI aus den Elementen ist  .
Beim Haber-Bosch-Verfahren ist
.
Beim Haber-Bosch-Verfahren ist  .
.
Alternativ ist es oft zweckmäßig, die Zusammensetzung der Gasphase über Molenbrüche
(Stoffmengenanteile)  anzugeben. In diesem Fall wird eine auf die Stoffmengenanteile bezogene
Gleichgewichtskonstante
anzugeben. In diesem Fall wird eine auf die Stoffmengenanteile bezogene
Gleichgewichtskonstante  erhalten:
erhalten:
 mit
mit 
Allgemein gilt hier:

Anwendungen des Massenwirkungsgesetzes
Für eine Vielzahl von Spezialfällen definiert das Massenwirkungsgesetzes – teilweise in vereinfachter Form – Gleichgewichtskonstanten für spezifische Reaktionsszenarien. So beschreiben Assoziations- und Dissoziationskonstanten das Gleichgewicht für Assoziations- und Dissoziationsprozesse. Das Löslichkeitsprodukt definiert die Gleichgewichtslöslichkeit von Salzen in Wasser. Komplexbildungskonstanten quantifizieren die Stabilität von Komplexverbindungen. Ionenprodukte werden durch Vereinfachung des Massenwirkungsgesetzes für elektrolytische Dissoziationsprozesse erhalten. Die quantitative thermodynamische Beschreibung der Säure-Base-Chemie durch Säurekonstanten und Basenkonstanten basiert auf dem Massenwirkungsgesetz. Die Protolyse von Essigsäure in wässeriger Lösung wird beispielsweise durch folgende stöchiometrische Reaktionsgleichung beschrieben:

Die Säurekonstante  von Essigsäure ergibt sich gemäß:
von Essigsäure ergibt sich gemäß:

Literatur
- Peter W. Atkins, Julio de Paula: Physikalische Chemie. Aus dem Englischen von Michael Bär, Anna Schleitzer und Carsten Heinisch. 5. Auflage. Wiley-VCH, Weinheim 2013, ISBN 978-3-527-33247-2.
- Andreas Heintz: Thermodynamik der Mischungen – Mischphasen, Grenzflächen, Reaktionen, Elektrochemie, äußere Kraftfelder. Springer-Verlag, Berlin/Heidelberg 2017, ISBN 978-3-662-49923-8, doi:10.1007/978-3-662-49924-5.
- Charles E. Mortimer, Ulrich Müller: Chemie – Das Basiswissen der Chemie. 13. Auflage. Georg Thieme Verlag, Stuttgart 2019, ISBN 978-3-132-42274-2.


© biancahoegel.de
Datum der letzten Änderung: Jena, den: 17.12. 2025