Carbene
Als Carbene bezeichnet man in der Chemie eine Gruppe von äußerst instabilen Verbindungen des zweiwertigen Kohlenstoffs mit einem Elektronensextett. Als Elektronenmangelverbindungen mit zwei nichtbindenden Elektronen am Kohlenstoff weisen sie eine hohe Reaktivität auf. Sie waren früher fast nur als Zwischenstufen bei chemischen Reaktionen bekannt, die meist sofort weiter reagieren. Das änderte sich mit der Entdeckung stabiler Carbene ab Ende der 1980er Jahre (besonders N-heterocyclische Carbene), was zu einem großen Aufschwung in der Forschung zu Carbenen führte und Anwendungen zum Beispiel in der Katalyse. Carbene gehören zu den Tetrylenen.
Geschichte
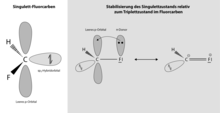
Die Geschichte der Carbene begann in den 1830er Jahren (Jean-Baptiste Dumas, Eugène-Melchior Péligot), als man erfolglos versuchte Methanol zu Methylen zu dehydrieren. Damals war noch nicht klar, dass Kohlenstoff vierwertig war. Zwischen 1892 und 1904 verfolgte John Ulric Nef von der Universität Chicago die Idee, dass Kohlenstoff manchmal zweiwertig wäre. 1897 sagte er die Existenz des Methylens voraus und behauptete es in wenigen Monaten synthetisieren zu können, konnte aber weder hier noch bei anderen Kohlenstoffverbindungen experimentelle Nachweise für seine Hypothese erbringen. Die hohe Reaktivität kurzlebiger Carbene aus Diazoverbindungen oder Ketenen wurde 1885 von Eduard Buchner und Theodor Curtius und 1912 von Hermann Staudinger demonstriert. Arbeiten über Carbene als Übergangszustände waren in den 1940er und 1950er Jahren populär, zum Beispiel in den Arbeiten von William von Eggers Doering, der Cyclopropane durch Reaktion von Carbenen mit Kohlenstoff-Doppelbindungen synthetisierte, und Philip Skell. Von Doering, Saul Winstein und Robert B. Woodward stammt auch der Name Carben. Doering und Prinzbach benutzten Carbene auch zur C-H Insertion. Ende der 1950er Jahre entdeckten Ronald Breslow und Hans-Werner Wanzlick dass Carbene durch Aminogruppen stabilisiert werden können, ohne dass ihnen die Isolation gelang. In den 1960er Jahren konnten Carbene schließlich spektroskopisch nachgewiesen und charakterisiert werden, was letzte Zweifel an ihrer Existenz ausräumte. So gelang Gerhard Herzberg 1959 der spektroskopische Nachweis von Methylen mit Blitzlichtphotolyse von Diazomethan. Herzberg zeigte auch aus den Spektren, dass es zwei Isomere gibt, was theoretisch den zwei quantenmechanischen Möglichkeiten eines Triplett- und Singulettzustands entspricht je nach gegenseitiger Spin-Ausrichtung der beiden ungepaarten Elektronen. Direkt nachgewiesen wurde das in den 1960er Jahren durch ESR-Spektroskopie an Aryl- und Diarylcarbenen, die durch Photolyse ihrer Diazoderivate bei tiefen Temperaturen (−190 Grad Celsius) in glasartig erstarrten Lösungsmitteln erzeugt wurden. In den 1960er Jahren gelang auch die Analyse von Carben-Reaktionen in der Gasphase. 1988 gelang Guy Bertrand die Isolierung eines stabilen Carbens, gefolgt 1991 von Anthony J. Arduengo mit den N-heterocyclischen Carbenen (NHC), die in kristalliner Form vorlagen und den eigentlichen Durchbruch brachten mit einer Fülle von Veröffentlichungen und vielen neuen stabilen Carbenen, die auch als Liganden von Übergangsmetallkomplexen in der Katalyse Verwendung fanden (zuerst 1995) ebenso wie die Carbene selbst in der organischen Katalyse. Carbenkomplexe selbst waren schon länger bekannt (Lew Alexandrowitsch Tschugajew 1912, Ernst Otto Fischer 1964, die Fischer-Carbene). Stabile Carbene fanden auch zum Beispiel bei der Stabilisierung von Radikalen und Diradikaloiden Verwendung.
Struktur und Aufbau
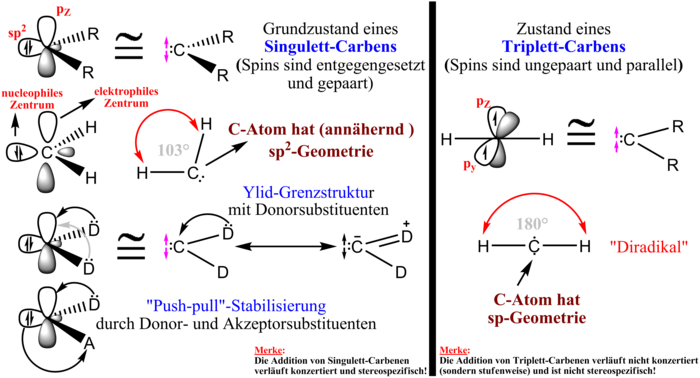
Man unterscheidet zwischen Singulett- und Triplett-Spinzuständen,
je nachdem, ob die beiden freien Elektronen
gepaart oder ungepaart (Triplett, dann handelt es sich um ein Diradikal) sind. Im Falle des
Singulettcarbens befinden sich sämtliche Elektronen in  -Hybridorbitalen,
am C-Atom bleibt ein unbesetztes
-Hybridorbitalen,
am C-Atom bleibt ein unbesetztes  z-Orbital
zurück. Das Triplettcarben trägt hingegen jeweils ein Elektron in einem
z-Orbital
zurück. Das Triplettcarben trägt hingegen jeweils ein Elektron in einem  -Orbital
und einem
-Orbital
und einem  -Orbital
(die
-Orbital
(die  -Bindung
wird über ein sp-Orbital hergestellt).
-Bindung
wird über ein sp-Orbital hergestellt).
Die Energiedifferenz zwischen Triplett- und Singulettzuständen ist meist
klein. Bestimmte Substituenten wie z. B.  -Donor-Substituenten
stabilisieren den Singulettzustand. Diese donieren Elektronendichte in das
unbesetzte p-Orbital am Carben-C-Atom und senken so dessen Elektrophilie.
Kombinierte starke -
und -Donorsubstituenten
können ein Carben im Singulettzustand stabilisieren und für eine nucleophile
Reaktivität sorgen. Umgekehrt verstärken stark elektronenziehende Substituenten
die Elektrophilie des Carbens zusätzlich. Singulett-Carbene lassen sich auch
durch sterische Einflüsse bevorzugen, da die Triplett-sp-Hybridisierung (180°),
beispielsweise in einem Fünfring wie dem Arduengo-Carben, nicht erreicht werden
kann.
-Donor-Substituenten
stabilisieren den Singulettzustand. Diese donieren Elektronendichte in das
unbesetzte p-Orbital am Carben-C-Atom und senken so dessen Elektrophilie.
Kombinierte starke -
und -Donorsubstituenten
können ein Carben im Singulettzustand stabilisieren und für eine nucleophile
Reaktivität sorgen. Umgekehrt verstärken stark elektronenziehende Substituenten
die Elektrophilie des Carbens zusätzlich. Singulett-Carbene lassen sich auch
durch sterische Einflüsse bevorzugen, da die Triplett-sp-Hybridisierung (180°),
beispielsweise in einem Fünfring wie dem Arduengo-Carben, nicht erreicht werden
kann.
Der Grundzustand des Methylens ist der Triplettzustand, der Singulettzustand stellt den angeregten Zustand dar. Die Energiedifferenz beträgt dabei etwa 36 kJ/mol. Neuere Untersuchungen legen eine substituentenabhängige Differenz von 7,1–20 kJ/mol nahe.
Stabile Carbene

Der Gruppe um den amerikanischen Chemiker Anthony J. Arduengo III gelang es 1991 zum ersten Mal ein Carben als kristalline, beliebig lange lagerbare Substanz zu erzeugen (N-Heterocyclische Carbene, NHC). Die ersten stabilen Carbene synthetisierte schon drei Jahre vorher Guy Bertrand (1988). Die Arduengo-Carbene weisen elektronegative Atome mit freien Elektronenpaaren als Substituenten am Carbenzentrum auf. Einerseits donieren die freien Elektronenpaare in das unbesetzte p-Orbital des Carbenkohlenstoffatoms und reduzieren so dessen Elektrophilie (s.o.), andererseits wird durch den Elektronenzug der elektronegativen Substituenten die Nukleophilie des freien Elektronenpaares am C-Atom verringert. Besonders stabile Carbene erhält man durch Resonanzstabilisierung, wenn π-Systeme gebildet werden, die nach der Hückel-Regel Aromaten darstellen. Bei den meisten Arduengo-Carbenen sind es Stickstoffatome, die die π-Bindung ausbilden, es gibt aber auch Beispiele mit anderen Atomen (z. B. S, O und P oder ylidischen Kohlenstoffatome in Amino(ylid)substituierten Carbenen). Außerdem sind nicht zwingend zwei π-Donoren zur Stabilisierung von Carbenen nötig, wie die Arbeitsgruppe von Guy Bertrand 2005 durch die Synthese von stabilen cyclischen (Alkyl)(amino)carbenen (CAACs) zeigte.
Auch Kohlenstoffmonoxid (CO) und die Vertreter der Verbindungsklasse der Isonitrile (RNC) können als stabile Carbene oder zumindest Carben-analoge Moleküle angesehen werden. Sie enthalten nämlich ebenfalls zweiwertigen Kohlenstoff, nur dass hier anstelle von zwei Substituenten lediglich ein Substituent (O oder NC) über eine Mehrfachbindung angebunden ist. Auch hier entsteht eine zusätzliche π-Bindung, die zu einer Gesamtbindungsordnung von 3 führt.
Carbenkomplexe
Wegen des freien Elektronenpaares eignen sich Carbene für den Einsatz als Liganden in organometallischen Komplexverbindungen. Vereinfacht erfolgt die Bindung zum Metall nach dem Dewar-Chatt-Duncanson-Modell. Die spezielle Unterklasse der N-heterocyclischen Carbene (z. B. Arduengo-Carbene) sind wegen ihres ausgeprägten Sigma-Donorcharakters in der Anorganischen Chemie von großer Bedeutung. Eine weite Anwendung finden sie in der Katalyse, wo sie häufig eine vorteilhafte Alternative zu Phosphanen bilden (z. B. bei der Metathese von Alkenen).
Synthesen
Carbene sind in der Regel zu reaktiv, um isoliert zu werden, so dass sie üblicherweise in situ erzeugt werden. Unter bestimmten Bedingungen, wie z. B. der Einbettung in eine inerte Matrix bei tiefer Temperatur, konnte aber auch Methylen stabilisiert und spektroskopisch untersucht werden.
Aus Diazoverbindungen
Einen guten Zugang zu Carbenverbindungen bietet die Thermo- oder Photolyse von Diazoverbindungen. Diese spalten ein Stickstoffmolekül ab und erzeugen so ein Carben. Für niedrigmolekulare Diazoverbindungen stellt die unkontrollierte Zersetzung ein erhebliches Problem dar, weshalb auch diese oft in situ aus geeigneten Vorläufern wie z. B. Hydrazonen hergestellt werden. Substituierte Hydrazone, wie bspw. Tosylhydrazone können auch mit starken Basen zum Carben umgesetzt werden. Diese Reaktion ist als Bamford-Stevens-Reaktion bekannt.
Aus Ketenen
Auch Ketene können eingesetzt werden, welche ein Molekül Kohlenmonoxid abspalten. Ketene sind allerdings ihrerseits aufwendig zu synthetisieren und neigen zu Polymerisationen, sodass sie in der Praxis selten zur Erzeugung von Carbenen benutzt werden.
Durch α-Eliminierung
Die Untersuchung dieser Reaktion führte zur Entwicklung der Carbenchemie. Zunächst entfernt man mit einer starken Base das Proton von Chloroform. Das entstehende Trichlormethylanion geht eine α-Eliminierung ein, sodass Dichlorcarben entsteht. Mittlerweile wird die Reaktion mit Chloroform und Natronlauge unter Zuhilfenahme eines Phasentransferkatalysators durchgeführt. Als solcher fungiert im Allgemeinen ein quartäres Ammoniumsalz.
Weitere
Es existieren weitere Möglichkeiten zur Carbenerzeugung wie z. B. aus Yliden, Heterocyclen oder sterisch gehinderten Alkanen.
Reaktionen
Insertionen
Carbene können in C–H-Bindungen und C–X-Bindungen insertieren. Bei der Insertion in C-H-Bindungen muss wieder die Reaktivität der unterschiedlichen Spinzustände des Carbens berücksichtigt werden. Die Singulettform addiert in einem intermediären Schritt an die C–H-Bindung, sodass es zu einer Wanderung des H-Atoms an das Carben-C-Atom kommt. Es bildet sich eine neue C–C-Bindung unter Retention der Konfiguration des Substrates. Das Triplettcarben hingegen abstrahiert ein Wasserstoffatom vom Substrat und rekombiniert mit dem entstandenen Radikal. Somit ist die Reaktion nicht stereospezifisch, weil die Geometrie des intermediären Radikals nicht festgelegt ist.
Bertrand und seine Mitarbeiter konnten außerdem zeigen, dass cyclische (Alkyl)(amino)carbene in H–H-Bindungen insertieren können. Der Grund hierfür liegt in einem gegenüber NHCs energetisch höherem höchsten besetzten Molekülorbital (HOMO) und einem geringeren Singlet-Triplet-Abstand. Das führt zu einer Senkung der Aktivierungsenergie von etwa 150 kJ/mol auf etwa 100 kJ/mol.
Cycloadditionen
An Alkene
Eine der bekanntesten Reaktionen ist die Addition von Carbenen an Alkene.
Sie führt zu Cyclopropanen. Dabei ist
die Addition eines Singulettcarbens immer stereospezifisch und verläuft unter
Erhaltung der Stereochemie
des Alkens. Ein Triplettcarben kann dagegen zu stereospezifischen Produkten
führen, muss aber nicht. Man erklärt dies durch einen konzertierten Mechanismus,
den das Singulettcarben eingeht. Das Triplettcarben addiert dagegen zunächst an
ein C-Atom im Alken. Der Ringschluss zum Cyclopropan ist spinverboten. Es muss
zunächst noch zu einer Spininversion kommen, welche Zeit benötigt. In diesem
Zeitraum ist daher eine Rotation um eine C-C-Bindungsachse möglich. Ob ein
Triplettcarben stereospezifisch addiert, hängt also davon ab, ob eines der Rotamere
energetisch bevorzugt wird.
Es zeigt sich, dass ein Carben die Doppelbindung nicht
direkt angreifen kann. Dieser Vorgang wäre bzgl. der Orbitalsymmetrie
symmetrieverboten. Es lassen sich keine Orbitale
der Reaktanten mit gleicher Symmetrie
zur Deckung bringen. Stattdessen muss das Carben seitlich, quasi mit einem
Lappen des -Orbitals
voran, angreifen. Gefahr droht bei Verwendung von Lösemitteln, die den
Singulettzustand zum Triplettzustand deaktivieren. Dann ist die Reaktion unter
Umständen nicht mehr stereospezifisch.
An Alkine
Diese Cycloaddition führt statt zu Cyclopropanen zu Cyclopropenen.
An Aromaten
Diese Additionen dienen unter anderem zur Ringerweiterung von cyclischen Aromaten. Es werden zumeist carbonylsubstituierte Carbene aufgrund ihrer stärkeren Elektrophilie verwendet. Zunächst addiert das Carben an eine formale Doppelbindung des Aromaten. Es entsteht ein 6-3-Bicyclus. Es kommt zu einer Umlagerung, wobei die beiden Ringen gemeinsame Bindung aufgelöst wird. Zurück bleibt ein, um eine C-Gruppe erweiterter, Siebenring.
Umlagerungen
Wie viele Elektronenmangelverbindungen sind auch Carbene zu Umlagerungen fähig. Durch 1,2-Verschiebungen von Substituenten des Carbens werden Alkene gebildet.
Organokatalyse
Neben den Einsatz als Liganden in Metall katalysierten Reaktionen können Carbene selbst als Organokatalysatoren verwendet werden. Benutzt werden dabei meist N-heterocyclische Carbene.
Siehe auch


© biancahoegel.de
Datum der letzten Änderung: Jena, den: 22.02. 2025