Aminosäurenstoffwechsel
| Übergeordnet |
| Carbonsäure-Metabolismus Stickstoffmetabolismus Amin-Stoffwechsel |
| Untergeordnet |
| schwefelhaltige A. Glutamin-Familie-A. Aspartat-Familie-A. Serin-Familie-A. Aromatische A. Histidin-Familie-A. nicht-proteinogene A. D-Aminosäuren |
| Gene Ontology |
|---|
|
|
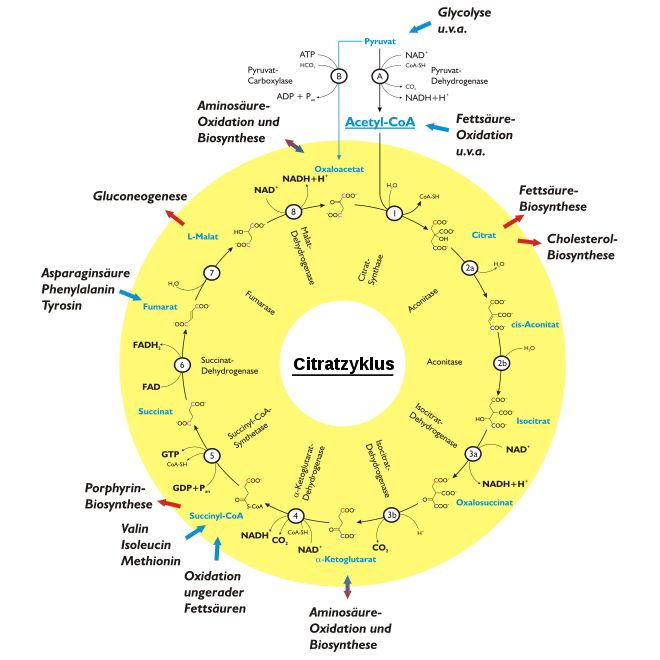
Als Aminosäurenstoffwechsel wird die Gesamtheit aller biochemischen Prozesse bezeichnet, die in einem Lebewesen zum Aufbau oder Abbau von Aminosäuren führen.
Biosynthese der proteinogenen L-Aminosäuren
Alanin wird aus Pyruvat gebildet. Glycin und Serin können aus 3-Phosphoglycerat, einem Intermediat der Glycolyse synthetisiert werden. Glutamat wird aus α-Ketoglutarat (Citratzyklus) gebildet. Aus Glutamat kann dann Prolin, Arginin und Glutamin produziert werden. Aus Oxalacetat (Citratzyklus) können Aspartat und Asparagin hergestellt werden. Cystein entsteht beim Abbau der schwefelhaltigen essentiellen Aminosäure Methionin. Tyrosin wird aus der essentiellen Aminosäure Phenylalanin generiert. Essentielle Aminosäuren können vom menschlichen Körper nicht gebildet werden, da ihre Synthese relativ umständlich ist und im Laufe der Evolution dafür notwendige Enzyme verloren gegangen sind. Das trifft beispielsweise auf die aromatischen Aminosäuren Phenylalanin und Tryptophan sowie auf die verzweigten Aminosäuren Valin, Leucin und Isoleucin zu.
Abbau der proteinogenen L-Aminosäuren
| Aminosäure | Ketogene Produkte | Glucogene Produkte |
|---|---|---|
| Alanin | Pyruvat | |
| Glycin → Serin | Pyruvat | |
| Threonin | Acetat | Pyruvat, Succinyl-CoA |
| Cystein | Pyruvat | |
| Asparagin → Aspartat | Oxalacetat / Fumarat | |
| Glutamin → Glutamat | α-Ketoglutarat | |
| Prolin → Glutamat | α-Ketoglutarat | |
| Arginin → Glutamat | α-Ketoglutarat | |
| Histidin → Glutamat | α-Ketoglutarat | |
| Methionin | Succinyl-CoA | |
| Lysin | Acetyl-CoA | |
| Phenylalanin → Tyrosin | Acetoacetat | Fumarat |
| Tryptophan | Acetyl-CoA | Pyruvat |
| Valin | Succinyl-CoA | |
| Leucin | Acetoacetat, Acetyl-CoA | |
| Isoleucin | Acetyl-CoA | Succinyl-CoA |
Die proteinogenen Aminosäuren werden auf vielfältige Weise abgebaut. Sie können zu Vorstufen für die Gluconeogenese abgebaut werden und sind somit glukogen. Werden sie zu Vorstufen der Ketonkörper-Synthese abgebaut so sind sie ketogen. In den meisten Fällen steht am Anfang des Aminosäurenabbaus die Abspaltung der Aminogruppe durch Desaminierung. Der Stickstoff wird über Glutamat und Aspartat in den Harnstoffzyklus eingeschleust und bei Säugetieren als Harnstoff über die Niere ausgeschieden. Daneben ist bei vielen Tierarten auch eine Ausscheidung über Harnsäure oder Ammoniumionen verbreitet.
Angeborene Aminosäureabbaudefekte
Fast alle werden autosomal-rezessiv vererbt und führen unbehandelt zu meist schweren und irreversiblen Schäden des zentralen Nervensystems. Die geläufigsten Formen sind:
- Phenylketonurie, PKU, syn.: Föllingsche Krankheit, der Tyrosinstoffwechsel ist gestört
- Albinismus, der Tyrosin- bzw. Dopastoffwechsel ist nur in den Melanozyten gestört, körperweit wäre letal
- Alkaptonurie, der Tyrosinstoffwechsel gestört
- Ahornsirupkrankheit syn.: Ketoacidurie, Leucin-, Isoleucin- und Valin-stoffwechsel sind gestört
Stoffwechselwege
Literatur
- J. M. Berg, J. L. Tymoczko, L. Stryer: Biochemie. 6. Auflage. Spektrum Akademischer Verlag, Elsevier GmbH, München 2007; ISBN 978-3-8274-1800-5.


© biancahoegel.de
Datum der letzten Änderung: Jena, den: 26.09. 2024