Mitochondrium
| Übergeordnet |
| Organell |
| Untergeordnet |
| Membran Intermembranraum Matrix Nukleoid Proteinkomplexe Mitoribosom Mitogenom (Chondriom) mtDNA |
| Gene Ontology |
|---|
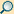
1. Nucleolus (Kernkörperchen)
2. Zellkern (Nukleus)
3. Ribosomen
4. Vesikel
5. Raues (Granuläres) ER (Ergastoplasma)
6. Golgi-Apparat
7. Cytoskelett
8. Glattes (Agranuläres) ER
9. Mitochondrien
10. Lysosom
11. Cytoplasma (mit Cytosol und Cytoskelett)
12. Peroxisomen
13. Zentriolen
14. Zellmembran
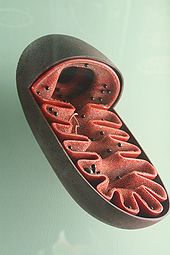
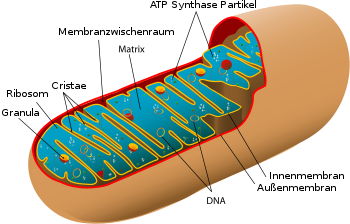
• Matrix
• Innen- und Außenmembran
• Membranzwischenraum (Intermembranraum),
• ATP-Synthase-Komplexen
• Cristae
• mitochondrialen Ribosomen (Mitoribosomen)
• Granula
• vielfach vorhandener zirkulärer mitochondrialer DNA (mtDNA)
Als Mitochondrium oder Mitochondrion (zu altgriechisch μίτος mitos ‚Faden‘ und χονδρίον chondrion ‚Körnchen‘; veraltet Chondriosom) wird ein Zellorganell bezeichnet, das von einer Doppelmembran umschlossen ist und eine eigene Erbsubstanz enthält, die mitochondriale DNA. Mitochondrien kommen als kugel- oder röhrenförmige Gebilde in den Zellen fast aller Eukaryoten vor, nicht aber bei Prokaryoten.
Mitochondrien regenerieren über die Atmungskette das energiereiche Molekül Adenosintriphosphat (ATP). Neben dieser oxidativen Phosphorylierung erfüllen sie weitere essentielle Aufgaben für die Zelle, beispielsweise sind sie an der Bildung von Eisen-Schwefel-Cluster beteiligt.
Allgemeines
Besonders viele Mitochondrien befinden sich in Zellen mit hohem Energieverbrauch; das sind unter anderem Muskelzellen, Nervenzellen, Sinneszellen und Eizellen. In Herzmuskelzellen erreicht der Volumenanteil von Mitochondrien 36 %. Sie haben einen Durchmesser von etwa 0,5–1,5 µm und sehr unterschiedliche Formen, von Kugeln bis zu komplexen Netzwerken. Mitochondrien vermehren sich durch Wachstum und Sprossung, die Anzahl von Mitochondrien wird dem Energiebedarf der Zelle angepasst. Eukaryotische Zellen, die ihre Mitochondrien verlieren, können diese nicht mehr regenerieren. Es gibt auch Eukaryoten ohne Mitochondrien, z.B. einige Protozoen. Die Anzahl der Mitochondrien pro Zelle liegt typischerweise bei einer Größenordnung von 1000 bis 2000 bei einem Volumenanteil von 25 %, jedoch können diese Werte je nach Zelltyp und Organismus sehr stark variieren. So liegt die Anzahl in der reifen Spermazelle des Menschen bei etwa vier bis fünf im Mittelstück („Hals“) gelegenen Mitochondrien, bei der reifen Eizelle dagegen bei mehreren hunderttausend.
Mitochondrien werden über das Plasma der Eizelle in der Regel nur von der Mutter vererbt, was Anlass zur Erforschung mütterlicher Verwandtschaftslinien (Matrilinien) war. Krankheiten, die durch Mutationen in mitochondrialen Genen verursacht werden, werden also normalerweise auch nur von der Mutter weitervererbt.
Mittlerweile hat sich herausgestellt, dass auch durch das Spermium einige männliche Mitochondrien in das Plasma der befruchteten Eizelle (Zygote) importiert werden. Diese „männlichen“ Mitochondrien werden aber normalerweise recht schnell eliminiert. Es gibt jedoch einige wenige Fälle, in denen Mediziner nachweisen konnten, dass die Mitochondrien des Kindes aus der väterlichen Linie stammten.
Bislang sind etwa 50 Krankheiten (Mitochondriopathien) bekannt, die durch mitochondriale Fehlfunktionen hervorgerufen werden können.
Der deutsche Pathologe und Histologe Richard Altmann entdeckte das Mitochondrium 1886. Die populärwissenschaftlich oft verwendete Bezeichnung „Kraftwerk der Zelle“ (englisch powerhouse of the cell) für das Mitochondrium wurde 1957 von Philip Siekevitz geprägt.
Aufbau
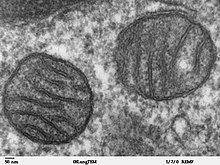
Die Hülle der Mitochondrien besteht aus einer äußeren und einer inneren Membran, die aus Phospholipid-Doppelschichten und Proteinen aufgebaut sind. Beide Membranen unterscheiden sich in ihren Eigenschaften. Durch diese Membranen existieren vier unterschiedliche Kompartimente: Die äußere Membran, der Intermembranraum (der Raum zwischen den beiden Membranen), die innere Membran/die Cristae und die Matrix (der Raum innerhalb der inneren Membran).
Mitochondrien sind in der Lage, sich (durch Fusion) zu verbinden und (durch Fission) wieder zu teilen; bei Hefezellen laufen pro Minute etwa zwei Mitochondrienfusionen bzw. -fissionen ab. Daher ist keine genaue Bestimmung der aktuellen Mitochondrienanzahl einer Zelle möglich.
Jedoch treten Mitochondrien nicht nur als separate, bohnenförmige Organellen auf. Stattdessen können sie auch tubuläre, also röhrenförmige, und teilweise verzweigte Netzwerke bilden. Form und Funktion stehen hierbei in einer engen Beziehung. Sie unterliegen vielfältigen Einflüssen und einer dynamischen Regulation. Solche mikrotubulären Extensionen, die größer als die T-Tubuli in Skelettmuskeln sind, und die über teilweise größere Distanzen Mitochondrien miteinander verbinden, ermöglichen einen intermitochondrialen Elektrolytaustausch und eine Stabilisierung des Membranpotentials. Sie werden vor allem bei mitochondrialen Myopathien beobachtet, können aber viel seltener auch in normalen Muskelzellen auftreten. Möglicherweise dienen sie dem Überleben der Mitochondrien unter Stress, wie bei der Stress-induzierten Mitophagie.
Außenmembran
Die äußere Membran umschließt das gesamte Mitochondrium und enthält Kanäle aus Proteinkomplexen, welche den Austausch von Molekülen und Ionen zwischen dem Mitochondrium und dem Cytosol ermöglichen. Große Moleküle können die Membran nicht passieren.
Die mitochondriale Außenmembran, die das gesamte Organell umschließt und nicht gefaltet ist, besitzt ein Gewichtsverhältnis von Phospholipid zu Protein von 1:1 und ist damit der eukaryotischen Plasmamembran ähnlich. Sie enthält zahlreiche integrale Proteine, die Porine. Porine bilden Kanäle, die freie Diffusion von Molekülen mit einer Masse von bis zu 5000 Dalton durch die Membran ermöglichen. Größere Proteine können in die Mitochondrien eindringen, wenn eine Signalsequenz an ihrem N-Terminus an eine große Proteinuntereinheit der Translokase bindet, von der sie dann aktiv durch die Membran bewegt werden. Treten Risse in der äußeren Membran auf, so können Proteine aus dem Intermembranraum ins Cytosol austreten, was zum Zelltod führen kann. Die mitochondriale Außenmembran kann sich mit der des Endoplasmatischen Retikulum (ER) zusammenschließen. Diese mitochondrien-assoziierten Membranstrukturen (MAM, englisch mitochondria-associated ER-membrane) sind wichtig für den Signalaustausch zwischen ER und Mitochondrium und spielen eine Rolle beim Lipidtransfer. Die Fusion wird katalysiert durch Mitofusine (MFN). Dabei handelt es sich um GTPasen der Dynamin-Superfamilie. Bei Säugetieren sind die Mitofusine MFN1 und MFN2 weit verbreitet. Beide Proteine haben zu 80 % eine identische Sequenz. Mutationen von MFN2 können beim Menschen die neurodegenerative Erkrankung Morbus Charcot-Marie-Tooth Typ 2A (CMT2A) hervorrufen.
Intermembranraum
Der Intermembranraum (Membranzwischenraum) ist der Raum zwischen der äußeren Membran und der inneren Membran. Da die äußere Membran frei durchlässig für kleine Moleküle ist, ist die Konzentrationen von kleinen Molekülen wie Ionen und Zuckern im Intermembranraum identisch mit der im Cytosol. Große Proteine allerdings benötigen eine spezifische Signalsequenz, um durch die Membran transportiert zu werden, sodass sich die Zusammensetzung der Proteine zwischen Intermembranraum und Cytosol unterscheidet. Ein Protein, das auf diese Art im Intermembranraum gehalten wird, ist Cytochrom c.
Innere Membran
Die Proteine der inneren Membran lassen sich nach ihrer Funktion in fünf Gruppen einteilen:
- Proteine der Komplexe für den Elektronentransport der Atmungskette, die Redox-Reaktionen durchführen
- ATP-Synthasen für die Synthese von ATP in der Matrix
- Transport-Proteine für den Eintritt und Austritt von Metaboliten
- Proteine der Membrankomplexe für den Einbau und Transport von Proteinen
- Proteine für die Verschmelzung (Fusion) und für die Teilung (Fission) von Mitochondrien
Die innere Membran enthält die verschiedenen Proteinkomplexe der Atmungskette für die oxydative Phosphorylierung und hat ein Gewichtsverhältnis von Phospholipid zu Protein von 1:3. Mehr als 150 verschiedene Arten von Polypeptiden und rund ein Fünftel aller Proteine eines Mitochondriums sind in der inneren Membran lokalisiert. Ihre lipidhaltige Doppelschicht ist reich an Cardiolipin. Dieses 1942 zuerst in Rinderherzen entdeckte Phospholipid ist kennzeichnend für mitochondriale und bakterielle Plasmamembranen und so auch in Prokaryoten zu finden. Cardiolipin enthält vier Fettsäuren anstelle von sonst zwei und trägt zur Ionenundurchlässigkeit der Membran bei. Im Gegensatz zur äußeren Membran enthält die innere Membran keine Porine und ist für wasserlösliche Moleküle nahezu undurchlässig. Fast alle Ionen und Moleküle benötigen daher für den Durchtritt spezielle Membrantransporter. Proteine werden mittels Oxa1 in die innere Membran transportiert und via der Translokase der inneren Membran (TIM) in die Matrix. Darüber hinaus existiert zwischen dem Intermembranraum und der Matrix ein Membranpotential, aufgebaut durch die Enzymaktivität der Atmungskette.
Die innere Membran umschließt den internen Flüssigkeitsraum des Mitochondriums, dessen Matrix. Diese entspricht dem Cytosol von Bakterien und enthält die mitochondriale DNA (mtDNA), die Enzyme des Citratzyklus und eigene mitochondriale Ribosomen (Mitoribosomen).
Aufbau der inneren Membran
Der Cristae-Typ (von lat. crista „Kamm“) besitzt an der inneren Membran zahlreiche Einstülpungen, die Cristae. Dadurch wird die Oberfläche der inneren Membran, an der chemische Reaktionen stattfinden können, erheblich vergrößert und ihre Fähigkeit, ATP zu produzieren, erhöht. Die innere Membran enthält große Proteinkomplexe der Atmungskette, welche für die Energiegewinnung zuständig sind. Der andere Mitochondrien-Typ heißt Tubuli-Typ und findet sich beispielsweise in steroidproduzierenden Zellen; bei ihm bildet die innere Membran Röhren aus.
Bei schlauchförmigen Einstülpungen mit perlenartigen runden Aussackungen spricht man vom Sacculi-Typ. Bei Mitochondrien einer Leberzelle ist die Fläche der inneren Membran etwa fünfmal größer als die der äußeren Membran. Dieses Verhältnis ist variabel, und Mitochondrien aus Zellen mit erhöhtem Bedarf an ATP, wie Muskelzellen, enthalten noch mehr Cristae. Die Cristae sind an der Innenseite mit kleinen runden Körpern mit einem Durchmesser von 8,5 nm, besetzt, die als F1-Partikel, Elementarpartikel oder ATP-Synthase-Partikel bekannt sind. Hier findet im Verlauf der Zellatmung die ATP-Bildung statt. Die Einstülpungen oder Falten der inneren Membran sind nicht zufällig und können ihre chemiosmotische Funktion beeinträchtigen.
Mitoplast
Mitoplast ist eine Bezeichnung für das Mitochondrium ohne Außenmembran: ein Mitochondrium, dessen Außenmembran (für Untersuchungszwecke) entfernt wurde, so dass nur noch die intakte Innenmembran nebst Inhalt (Matrix) vorhanden ist.
Matrix
Der durch die innere Membran umschlossene Raum wird Matrix genannt. In ihm sind ⅔ aller Proteine eines Mitochondriums enthalten. Die Matrix ist wichtig bei der ATP-Produktion, die mit Hilfe der ATP-Synthase stattfindet. Die Matrix enthält eine hochkonzentrierte Mischung aus Hunderten von Enzymen sowie die speziellen mitochondrialen Ribosomen (Mitoribosomen), tRNA und mehrere Kopien des mitochondrialen Genoms (Mitogenoms). Außerdem ist in ihm die Konzentration an Intermediaten (Zwischenprodukten) des Citratzyklus der Beta-Oxidation sehr hoch. Zu den Hauptaufgaben der Enzyme gehört die Oxidation von Pyruvat und Fettsäuren sowie der Citratzyklus.
Da Mitochondrien eigenes genetisches Material besitzen, können sie selbst RNA und Proteine herstellen (Transkription und Translation). Es wurde gezeigt, dass in der mitochondrialen DNA-Sequenz (Mitogenom) 16.569 Basenpaare insgesamt 37 Gene codieren, davon sind 22 tRNA-, 2 rRNA- und 13 Peptid-Gene. Die 13 mitochondrialen Peptide sind in die innere Mitochondrienmembran integriert, zusammen mit Proteinen, die im Genom der Wirtszelle codiert sind und von ihr gebildet und dann eingeschleust werden.
Die mitochondrialen Ribosomen (Mitoribosomen) unterscheiden sich von den eukaryotischen Ribosomen im Cytosol. Die Unterschiede zu bakteriellen Ribosomen sind vergleichsweise gering, aber vorhanden. Mitochondriale Ribosomen beim Menschen sind etwa 55S Svedberg-Einheiten groß, gegenüber 70S bei Bakterien und 80S bei den cytosolischen Ribosomen. Es gibt zwar auch mitochondriale 70S-Ribosomen, aber nicht bei Säugern (Mammalia) einschließlich des Menschen. Der Intermembranraum zwischen den beiden Membranen enthält Enzyme, die Nukleotide unter ATP-Verbrauch phosphorylieren können.
Mitochondrien-assoziierte ER-Membran (MAM)
Die Mitochondrien-assoziierte ER-Membran (MAM, englisch mitochondria-associated ER membrane) ist ein weiteres strukturelles Element, dessen entscheidende Rolle in der zellulären Physiologie und Homöostase zunehmend erkannt wird. Während man einst vermutete, dass es nur eine permanent auftauchende Verunreinigung durch technische Schwierigkeiten bei der Fraktionierung sei, wurde es jetzt als membranöse Struktur an der Schnittstelle zwischen Mitochondrien und dem ER identifiziert. Eine physikalische Kopplung zwischen diesen zwei Organellen war bereits zuvor bei elektronenmikroskopischen Aufnahmen und in neuerer Zeit mit Fluoreszenz-Mikroskopie untersucht worden. Solche Untersuchungen ergaben die Vermutung, dass an der MAM, das bis zu 20 % der äußeren mitochondrialen Membran umschließt, ER und Mitochondrium nur noch durch eine 10–25 nm große Lücke getrennt sind und beide durch einen Proteinkomplex zusammengehalten werden.
Gereinigte MAM aus subzellulären Fraktionierungen hat gezeigt, dass es neben Ca2+-Ionenkanälen auch mit Enzymen angereichert ist, die in den Phospholipidaustausch involviert sind. Diese Hinweise auf eine besondere Rolle der MAM in der Regulation der Fettspeicherung und Signalübertragung haben sich bestätigt, mit bedeutenden Auswirkungen für mitochondrial-assoziierte zelluläre Phänomene, wie nachfolgend diskutiert. MAM hat nicht nur einen Einblick in die zugrunde liegenden mechanistischen Grundlagen von physiologischen Prozessen wie der intrinsische Apoptose und der Ausbreitung von Calcium-Signalen gegeben, sondern es verfeinerte auch unsere Sicht auf die Mitochondrien. Obwohl sie oft als statische und isolierte „Kraftwerke der Zelle“ gesehen werden, unterstreicht die Entwicklung der MAM, inwieweit Mitochondrien in die Zellphysiologie integriert wurden, mit enger physikalischer und funktioneller Kupplung ans Endomembransystem.
Phospholipid-Transfer
Die MAM ist angereichert mit Enzymen, die in der Lipid-Biosynthese involviert sind, wie z.B. Phosphatidylserin-Synthase auf der ER-Oberfläche und Phosphatidylserin-Decarboxylase auf der mitochondrialen Oberfläche. Da Mitochondrien als dynamische Organellen ständig Spaltung und Fusion durchlaufen, müssen sie für die Membranintegrität konstant und ausreichend reguliert mit Phospholipiden versorgt werden. Allerdings sind Mitochondrien nicht nur ein Ziel für Phospholipide, sondern spielen auch eine Rolle beim Austausch zwischen Organellen von (Zwischen-)Produkten des Phospholipid-Biosynthesewege, des Ceramid- und Cholesterinstoffwechsels sowie Glykosphingolipid-Anabolismus.
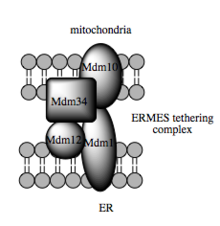
Die Transportkapazität dieser Stoffe ist von der MAM abhängig, von der gezeigt worden ist, dass sie den Transfer von Lipidzwischenprodukten zwischen Organellen erleichtert. Im Gegensatz zum Standard-Vesikel-Mechanismus des Lipidtransfers gibt es Hinweise, dass die räumliche Nähe des ER zur Mitochondrienmembran beim MAM das Flippen zwischen gegenüberliegenden Lipiddoppelschichten ermöglicht. Trotz dieses ungewöhnlichen und scheinbar energetisch ungünstigen Mechanismus, erfordert solch ein Transport kein ATP. Stattdessen wurde eine Abhängigkeit von einem Multiproteinkomplex gezeigt, der ERMES (englisch ER-mitochondria encounter structure) genannt wird, auch wenn unklar bleibt, ob dieser Komplex den Lipidtransfer direkt vermittelt oder aber erforderlich ist, um die Membranen in ausreichender Nähe zueinander zu halten, um so die Energiebarriere für ein direktes Lipid-Flipping zu senken.
MAM könnte auch Teil des sekretorischen Mechanismus sein, zusätzlich zu seiner Rolle beim intrazellulären Lipid-Stoffaustausch. Speziell scheint MAM eine Zwischenstation zwischen dem rauen ER und dem Golgi-Apparat zu sein, in dem Stoffwechselweg, der zu Very Low Density Lipoproteinen (VLDL) führt. Die MAM dient somit als entscheidender Stoffwechsel- und Umschlagplatz im Fettstoffwechsel.
Calcium-Signalgebung
Die bedeutende Rolle des ER für die Calcium-Signalgebung war lange vor der des Mitochondriums anerkannt; zum Teil, da die geringe Affinität der Ca2+-Kanäle in der äußeren Membran der angeblichen Verantwortlichkeit des Organells für intrazelluläre Ca2+-Ströme völlig widersprach. Durch die Anwesenheit des MAM wird dieser scheinbare Widerspruch aufgelöst: Die enge räumliche Verbindung zwischen den beiden Organellen resultiert in Ca2+-Mikrodomänen an Berührungspunkten, die eine effiziente Ca2+-Übertragung vom ER zu den Mitochondrien erleichtern. Die Übertragung erfolgt in Reaktion auf sogenannte „Ca2+-puffs“, erzeugt durch spontane Zusammenlagerung und Aktivierung von IP3R, einem anerkannten ER-Membran Ca2+-Kanal.
Perspektive
Die MAM ist von entscheidender Bedeutung für den Austausch von Information und Stoffwechselprodukten in der Zelle, die eine Vernetzung der Physiologie von ER und Mitochondrien ermöglicht. Zwischen beiden besteht nicht nur eine strukturelle, sondern auch eine funktionelle Kopplung, die entscheidend für die gesamte zelluläre Physiologie und Homöostase ist. Die MAM ermöglicht damit eine Sichtweise auf die Mitochondrien, die von der traditionellen Sichtweise dieser Organellen als eine statische, isolierte Einheit abweicht. Stattdessen wird die Integration der Mitochondrien in verschiedene zelluläre Prozesse betont, als das Resultat eines endosymbiontischen Vorgangs.
Funktion
- Wichtige Abbauwege: Citratzyklus, hierzu wird Pyruvat aus dem Cytosol in die Mitochondrienmatrix eingeschleust. Durch die Pyruvat-Dehydrogenase wird dann Pyruvat zu Acetyl-CoA decarboxyliert. Eine andere Quelle des Acetyl-CoA ist der Fettsäureabbau (β-Oxidation), welcher in tierischen Zellen in Mitochondrien stattfindet, in pflanzlichen Zellen jedoch nur in den Glyoxysomen und den Peroxisomen. Hierzu wird Acyl-CoA aus dem Cytosol über Bindung an Carnitin durch die innere Mitochondrienmembran geschleust und zu Acetyl-CoA umgesetzt. Aus Acetyl-CoA wird im Citratzyklus (auch Krebs-Zyklus oder Tricarbonsäure-Zyklus genannt) der überwiegende Teil der Reduktionsäquivalente (NADH+H+, FADH2) gewonnen, die dann innerhalb der Atmungskette in ATP umgewandelt werden.
- Atmungskette: Dabei wird mit Hilfe von Elektronen-Transportvorgängen und durch Anreicherung von Protonen ein elektrochemischer Gradient zwischen dem Intermembranraum und der mitochondrialen Matrix aufgebaut, der dazu dient, mittels der ATP-Synthase, ATP herzustellen (siehe chemiosmotische Kopplung). Die zum Aufbau des Gradienten benötigten Elektronen und Protonen werden durch oxidativen Abbau aus den vom Organismus aufgenommenen Nährstoffen (z.B. Glucose) gewonnen. Zunächst läuft im Zytoplasma die Glykolyse ab.
- Apoptose (Programmierter Zelltod)
- Calcium-Speicher: durch die Fähigkeit Calciumionen aufzunehmen und später wieder abzugeben, greifen Mitochondrien in die Calcium-Homöostase der Zelle ein.
- Synthese von Eisen-Schwefel-Clustern, die unter anderem von vielen Enzymen der Atmungskette benötigt werden. Diese Funktion wird inzwischen als die essentielle Funktion der Mitochondrien angesehen, d.h. als der Grund, warum fast alle eukaryotischen Zellen zum Überleben auf Mitochondrien angewiesen sind.
- Einzelne Schritte aus dem Harnstoffzyklus finden ebenfalls in Mitochondrien statt.
Evolution
Ursprung
Nach der Endosymbiontentheorie geht man davon aus, dass die Mitochondrien aus einer Symbiose von aeroben Bakterien (aus der Gruppe der α-Proteobakterien, Gattung Rickettsia) mit den Vorläufern der heutigen Eukaryoten hervorgegangen sind. Ein alternativer Vorschlag ist die Aufnahme eines fakultativen anaeroben Bakteriums (Symbiont) durch ein methanogenes Archaeo (Wirt). Hinweise auf eine wie auch immer gestaltete Endosymbiose sind der Besitz eigener genetischer Information (mtDNA, Mitogenom oder – seltener – Chondriom), eine eigene Proteinsynthese – mit eigenen Ribosomen (Mitoribosomen) und eigenen tRNAs – und das Vorhandensein einer inneren Membran, die sich deutlich vom Bau der äußeren Membran unterscheidet und die der Synthese von ATP aus ADP dient. Die Mitochondrien sind jedoch so spezialisiert, dass sie allein nicht lebensfähig sind. Sie sind relativ eng mit anderen, seltener auftretenden Organellen, den Hydrogenosomen, verwandt. Diesen fehlt jedoch meist – nicht immer – eigene DNA, genauso wie den ebenfalls verwandten Mitosomen.
MROs
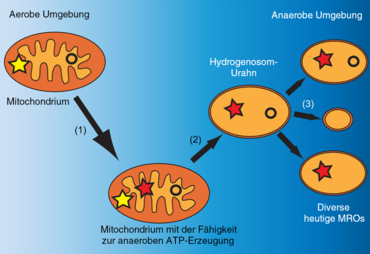
(1) Erwerb von Enzymen zur anaeroben Energiegewinnung.
(2) Verlust der Fähigkeit zur oxidativen Phosphorylierung.
(3) Verlust verschiedener mitochondrialer Funktionen.
Gelbe Sternchen stehen für die Elektronentransportkette, während rote Sternchen den hydrogenosomalen anaeroben ATP-Syntheseweg darstellen. Die Kreise stellen die mitochondrialen Genome dar.
Zusammen mit den Hydrogenosomen und Mitosomen werden Mitochondrien daher als „mitochondrienverwandte Organellen“ (englisch mitochondrion-related organelles, MROs) oder „mitochondrienähnliche Organellen“ (englisch mitochondrion-like organelles, MLOs) klassifiziert. Zu diesen gehören auch die anaeroben und DNA-freien Organellen von Henneguya salminicola (alias H. zschokkei, Myxozoa). Auch die Einzellergattung Blastocystis besitzt MLOs mit eigenem Genom.
Alle Eukaryoten besitzen fast ausnahmslos MROs, also Mitochondrien oder Organellen von einem dieser verwandten Typen. Eine Ausnahme ist Monocercomonoides (Excavata). Man nimmt an, dass diese Einzeller durch horizontalen Gentransfer ein zytosolisches System erworben hatten, um für die Proteinsynthese erforderliche Eisen-Schwefel-Cluster bereitzustellen. Danach waren ihre mitochondrialen Organellen in all ihren Funktionen überflüssig und gingen verloren.
Eine im Herbst 2020 veröffentlichte Studie legt anhand von umfangreichen Genomanalysen nahe, dass – obwohl bisher noch keine primär amitochondrialen Eukaryoten gefunden wurden – die Vorfahren der Eukaryoten zuerst ihre komplexen Genome mit den zugehörigen Strukturen und danach die Mitochondrien (oder Vorläufer davon) erworben haben.
Genom
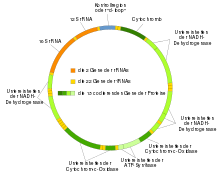
Die Mitochondrien besitzen (fast ausnahmslos) ein eigenes Genom (Chondriom, auch Mitogenom), das sich, häufig mehrfach kopiert, in der mitochondrialen Matrix befindet. Das Genom liegt gewöhnlich in einer einzigen zirkulären und doppelsträngigen DNA (mtDNA) vor (siehe auch Plasmid), die aber in mehreren (etwa zwei bis zehn) Kopien vorliegen kann. Die mtDNA besitzt einen eigenständigen Verdopplungszyklus. Mitochondrien werden als semiautonom bezeichnet, ihr Genom codiert selbst nur einen kleinen Teil der vom Mitochondrium benötigten Proteine. Bei Tieren besteht das Mitogenom typischerweise aus 37 Genen bei einer Länge von 16 kb (Basenpaaren). Beim Menschen kontrollieren die 37 mitochondrialen Gene die Synthese von 13 der ca. 80 Protein-Untereinheiten der Atmungskette, die restlichen 800–1000 verschiedenen mitochondrialen Proteine werden im Kerngenom kodiert.
Die nicht für Proteine codierenden Gene der mtDNA codieren für die rRNA und für alle benötigten tRNAs.
Veränderungen im Mitochondriengenom werden in der Forschung zur Aufklärung von Abstammungslinien der Arten, sowie verschiedener ethnischer Gruppen des Menschen genutzt, so etwa vom Genographic-Projekt.
Vermehrung
Mitochondrien werden nicht neu gebildet, sondern entstehen durch Wachstum und Sprossung. Der Großteil der mitochondrialen Proteine wird im Cytosol synthetisiert und anschließend in die Mitochondrien transportiert. Der Transport dieser Proteine in die Mitochondrien erfolgt über die äußere Membran durch den TOM-Komplex (englisch translocase of outer mitochondrial membrane) und über die innere Membran durch den TIM-Komplex (engl. translocase of inner mitochondrial membrane) und beinhaltet die Funktion von Chaperonen, besonders Hsp70.
Die Vermehrung der Mitochondrien ist über alle Arten der Eukaryoten homolog und stark konserviert.
Ausmaß und Zeitpunkt der Mitochondrien-Vermehrung hängen jeweils vom Bedarf ab. Bei der Zellteilung werden sie von der Mutterzelle auf die Tochterzellen verteilt.
Verbrauchte Mitochondrien werden mit Hilfe des Endoplasmatischen Retikulums, des Golgi-Apparats und der Lysosomen abgebaut.
Mitochondrien in komplexen Chloroplasten
Interessanterweise gibt es komplexe Chloroplasten (die nach der Endosymbiontentheorie aus einer sekundären Endosymbiose eines Eukaryoten mit einer Algenzelle – beispielsweise einer Rotalge – entstanden sind) mit eigenen Mitochondrien. Einige Dinoflagellaten wie Kryptoperidinium und Durinskia (beide Peridiniaceae, auch englisch dinotoms) haben einen von Diatomeen (Heterokontophyta) abgeleiteten Chloroplasten. Diese Chloroplasten sind von bis zu fünf Membranen umgeben, je nachdem, ob man den gesamten Diatomeen-Endosymbionten als den Chloroplasten ansieht oder nur die darin enthaltenene Rotalge als Chloroplast zählt. Der Diatomeenendosymbiont ist relativ wenig reduziert worden – er behält immer noch seine ursprünglichen Mitochondrien und verfügt über Endoplasmatisches Retikulum, eukaryotische Ribosomen, einen Zellkern und natürlich den von Rotalgen abstammenden komplexen (sekundären) Chloroplasten – praktisch eine vollständige Zelle – alles im Innern des Wirts.
Die Ocelloide der einzelligen Dinoflagellaten in der Familie Warnowiaceae (Warnowiiden) sind ebenfalls komplexe Organellen, zusammengesetzt aus Plastiden und Mitochondrien.
Mögliche Beziehungen zum Altern
Die Mitochondrien, als Kraftwerke der Zelle, können undichte Stellen in der Atmungskette aufweisen; d. h. bei der respiratorischen Oxidation kann es zur Freisetzung von Elektronen kommen. Diese können reaktive Sauerstoffspezies bilden, was oxidativen Stress verursachen kann, welcher zu einer hohen Mutationsrate der mitochondrialen DNA (mtDNA) führen kann. Diese hypothetischen Zusammenhänge zwischen Altern und oxidativem Stress sind nicht neu und wurden bereits vor über 50 Jahren vorgeschlagen. Sie werden in neueren Arbeiten angezweifelt.
Oxidativer Stress kann zu mitochondrialen DNA-Mutationen führen, welche enzymatische Anomalien verursachen können, was wiederum zu weiterem oxidativem Stress führen kann. Ein Teufelskreis ist möglich. Neuere Messungen haben jedoch ergeben, dass die Akkumulationsgeschwindigkeit von Mutationen in mitochondrialer DNA 1 Mutation pro 7884 Jahre beträgt (das heißt 10−7 bis 10−9 pro Base pro Jahr, die jüngsten gemeinsamen Vorfahren von Menschen und Affen betrachtend), und damit vergleichbar ist zu den Mutationsraten autosomaler DNA (10−8 Basen pro Generation).
Während des Alterns der Mitochondrien können verschiedene Änderungen auftreten. Gewebe von älteren Patienten zeigen eine Abnahme der enzymatischen Aktivität der Proteine der Atmungskette. Mutationen der mitochondrialen DNA können jedoch nur in 0,2 % der sehr alten Zellen gefunden werden. Es wird angenommen, dass große Deletionen im mitochondrialen Genom zu einem hohen Maß an oxidativem Stress und dem Absterben von Neuronen führen, als Auslöser der Parkinson-Krankheit.
Allerdings gibt es viele Diskussionen darüber, ob mitochondriale Veränderungen Ursachen des Alterns sind oder nur Merkmale des Alterns. Eine repräsentative Studie an Mäusen zeigte eine verkürzte Lebensdauer, jedoch keine Erhöhung der reaktiven Sauerstoffspezies trotz steigender mitochondrialer DNA-Mutationen. Es ist jedoch zu beachten, dass bei alternden Wildtypmäusen nicht so viele Mutationen in der mitochondrialen DNA akkumulieren wie bisher vermutet (und diese Mutationen einen geringeren Effekt haben als gedacht), was Zweifel an der Bedeutung von Mutationen in der mitochondrialen DNA für das natürliche Altern aufkommen lässt. Somit bleiben die genauen Zusammenhänge zwischen Mitochondrien, oxidativem Stress und Alterung ungeklärt.
Siehe auch
- Plastid, insbesondere Chloroplasten und komplexe Chloroplasten
Literatur
- Bruce Alberts u.a.: Molecular Biology of the Cell. 4. Auflage. 2002, ISBN 0-8153-3218-1.


© biancahoegel.de
Datum der letzten Änderung: Jena, den: 28.02. 2026