Lysosom
| Übergeordnet |
| Organell |
| Untergeordnet |
| Lysosomenmembran Lysosom-Glycocalyx Lumen Proteinkomplexe |
| Gene Ontology |
|---|
|
|
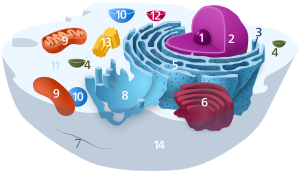
1. Nucleolus (Kernkörperchen)
2. Zellkern (Nukleus)
3. Ribosomen
4. Vesikel
5. Raues (Granuläres) ER (Ergastoplasma)
6. Golgi-Apparat
7. Cytoskelett
8. Glattes (Agranuläres) ER
9. Mitochondrien
10. Lysosom
11. Cytoplasma (mit Cytosol und Cytoskelett)
12. Peroxisomen
13. Zentriolen
14. Zellmembran
Lysosomen (von griechisch λύσις, von lysis ‚Lösung‘, und σῶμα sṓma ‚Körper‘) sind Zellorganellen in eukaryotischen Zellen. Es handelt sich dabei um von einer einfachen Biomembran umschlossene Vesikel mit saurem pH-Wert. Sie enthalten Verdauungsenzyme und werden teilweise im Golgi-Apparat gebildet. Die Funktion der Lysosomen besteht darin, Biopolymere in ihre Monomere zu zersetzen.
Aufbau
Lysosomen haben einen Durchmesser von 0,1–1,1 μm. Sie enthalten zur intrazellulären Verdauung von Material viele verschiedene hydrolysierende Enzyme wie Proteasen, Nukleasen und Lipasen. Diese Enzyme dienen der Hydrolyse von Proteinen, Polysacchariden, Nucleinsäuren und Lipiden, also aller wichtigen Gruppen von Makromolekülen. Diese erreichen nur eine hohe Aktivität in einer sauren Umgebung mit einem pH von 4,5–5. Dies dient dem Schutz der Zelle bei einem Aufbruch eines Lysosoms. In einem solchen Fall wären die Enzyme im pH-neutralen Milieu des Cytosols inaktiv. Dies ist ein Beispiel für die Wichtigkeit der Kompartimentierung innerhalb der Zelle. Der niedrige pH-Wert innerhalb der Lysosomen wird durch die Lysosomenmembran aufrecht gehalten. Lysosomen sind von einer Membran mit spezifischer Proteinausstattung umgeben. Eine V-Typ-ATPase transportiert pro ATP-Molekül zwei Protonen (2H+) in die Lysosomen. Die Membranproteine sind auf der Innenseite zum Schutz vor der Selbstverdauung stark glykosyliert.
Entstehung
Die hydrolytischen Enzyme und die Lysosomenmembran werden durch Ribosomen am rauen (granulären) Endoplasmatischen Retikulum (rER) gebildet und anschließend zum Golgi-Apparat transportiert. Die lysosomalen Enzyme erfahren im trans-Golgi-Apparat eine Sortierung und werden gezielt in Vesikel verpackt und zu den späten Endosomen transportiert. Im Fall der Hydrolasen ist ein spezifisches Signal bekannt: Mannose-6-phosphat-Gruppen (M6P) an ausschließlich Stickstoff-gekoppelten Oligosacchariden. Diese Modifikation findet im cis-Golgi-Apparat statt und wird von zwei Enzymen katalysiert: Eine Phosphotransferase erkennt, dass es sich um ein lysosomales Enzym handelt und heftet N-Acetylglucosamin-1-phosphat an ein oder zwei Mannosereste; das zweite Enzym schneidet den N-Acetylglucosamin-Rest ab, womit die Markierung durchgeführt ist.
Im trans-Golgi-Apparat werden die M6P-Reste von membranintegralen M6P-Rezeptoren erkannt. Im späten Endosom trennen sich die M6P-Rezeptoren bei pH 6 wieder von ihren Liganden und werden rezyklisiert.
Es gibt auch einen M6P unabhängigen Transportweg in die Lysosomen, z.B. bei den Membranproteinen der Lysosomen. Der Mechanismus ist nicht bekannt.
Aufgaben
.jpg)
mikroskopie
Verdauung zellfremden Materials
Lysosomen wirken auf mehrere Weisen an der Verdauung auf Zellebene mit. Durch Endozytose entstandene Endosomen verschmelzen mit primären Lysosomen zu sekundären Lysosomen. Bei Protisten wird dies Nahrungsvakuole genannt. In einigen Zelltypen werden Bruchstücke des im Lysosom verdauten zellfremden Materials in Form sogenannter Antigenfragmente durch MHC-II-Rezeptoren an der Zelloberfläche präsentiert. Dieser Vorgang spielt eine wichtige Rolle im Immunsystem. Als Beispiel für menschliche Zellen, die hierzu fähig sind, sind die Makrophagen zu nennen.
Verdauung zelleigenen Materials
Die Lysosomen verwerten nicht nur zellfremdes, sondern auch zelleigenes Material. Dies nennt man Autophagie. Hierbei werden Organellen oder Teile des Zytosols durch die Lysosomenenzyme zerlegt und wiederverwertet. Auf diese Weise erneuert sich die Zelle mit Hilfe der Lysosomen selbst. In einer menschlichen Leberzelle werden pro Woche die Hälfte aller Makromoleküle auf diese Weise zerlegt.
Programmierter Zelltod
Auch der programmierte Zelltod (Apoptose) durch eigene Lysosomenenzyme ist eine wichtige Aufgabe der Lysosomen. Durch Apoptose werden beispielsweise der Schwanz der Kaulquappe bei Amphibien oder die Schwimmhäute zwischen den Fingern des menschlichen Embryos abgebaut.
Erkrankungen mit lysosomaler Beteiligung
Ein Defekt in der Phosphotransferase führt zu einer so genannten lysosomalen Speicherkrankheit. Da keine Markierung mit Mannose-6-Phosphat stattfinden kann, werden die lysosomalen Enzyme nicht sortiert und gelangen unkontrolliert über die Plasmamembran in die extrazelluläre Matrix (I-Zellkrankheit, autosomal rezessiv vererbt). Andere lysosomale Speicherkrankheiten werden durch Defekte lysosomaler Hydrolasen ausgelöst. Dadurch kommt es zur Vermehrung von nicht-abgebautem Material in den Lysosomen (z.B. Morbus Hunter). Meistens sind schwere Krankheitserscheinungen die Folge. Bei einer Mutation der LIPA kommt es zur Lysosomalen saure Lipase-Defizienz. LIPA ist die lysosomale, saure Lipase, die wichtig für den Stoffwechsel von Cholesterin-Estern und Triglyzeriden ist.
Lysosomale Kumulation von Pharmaka
Schwache Basen mit lipophilen Eigenschaften neigen zur Akkumulation in sauren intrazellulären Kompartimenten wie den Lysosomen. Während die Plasma- und Lysosomenmembranen für die neutralen, ungeladenen Formen solcher Moleküle durchlässig sind, passieren die geladenen, protonierten Formen schwacher Basen die Membranen nicht und kumulieren in Lysosomen. Im Vergleich zum extrazellulären Bereich kann sich dadurch im Lysosom eine 100 bis 1000fach höhere Konzentration der schwachen Basen aufbauen. Dieser Mechanismus wird "Lysosomotropie" oder auch "acid trapping" genannt. Über ein zellbasiertes mathematisches Modell lässt sich die Kumulation lysosomotroper Substanzen berechnen.
Viele klinisch eingesetzte Medikamente sind schwache Basen und kumulieren in Lysosomen. Darüber lassen sich verschiedene pharmakologische Eigenschaften solcher Medikamente erklären. So liegt die Gewebekonzentration lysosomotroper Medikamente deutlich über der Plasmakonzentration; und die Halbwertszeit im Gewebe ist höher als im Plasma; als Beispiele seien hier Haloperidol, Levomepromazin oder Amantadin genannt. Zu den hohen Gewebekonzentrationen und langen Gewebehalbwertszeiten trägt neben der lysosomalen Akkumulation auch die Lipophilie der Substanzen und deren Absorption in fetthaltigen Gewebestrukturen bei. Wichtige lysosomale Enzyme, wie die saure Sphingomyelinase, können durch lysosomal akkumulierte Medikamente gehemmt werden. Solche Substanzen werden FIASMAs genannt. Dieses Akronym leitet sich von der englischen Bezeichnung Functional Inhibitor of Acid SphingoMyelinAse ab; zu dieser Substanzgruppe gehören z.B. Fluoxetin, Sertralin oder Amitriptylin.
Literatur
- Bruce Alberts u. a.: Molecular Biology of the Cell. 4. Auflage. Garland Science, New York 2002, ISBN 0-8153-4072-9.
- Neil A. Campbell, Jane B. Reece: Biologie. 6. Auflage. Spektrum, Heidelberg 2003, ISBN 3-8274-1352-4.


© biancahoegel.de
Datum der letzten Änderung: Jena, den: 13.01. 2026