Virushülle
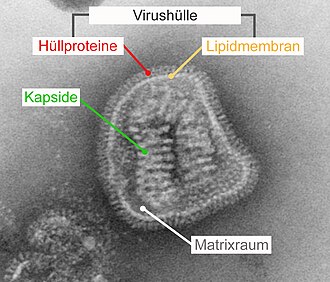
Die Virushülle (englisch viral envelope) ist eine bei bestimmten Viren vorhandene äußere Struktur, die aus Lipiden einer Phospholipid-Doppelschicht der ursprünglichen Wirtszelle und darin eingelagerten viralen Proteinen besteht. Die Virushülle umschließt meistens ein Kapsid, in das wiederum die virale Nukleinsäure verpackt ist. Je nach Virusart entsteht die Hülle aus der Zellmembran an der Zelloberfläche oder aus Membranen des Endoplasmatischen Retikulums (ER) bzw. Golgi-Apparates im Inneren der Zelle.
Das Vorhandensein einer Virushülle ist ein wichtiges Kriterium bei der Einteilung von Viren, der sogenannten Virus-Taxonomie. Dabei werden die behüllten Viren von den unbehüllten oder „nackten“ Viren abgegrenzt. Während unbehüllte Viren die infizierte Zelle stets durch Zerstörung der Wirtszelle verlassen müssen, können behüllte Viren ohne eine solche Lyse durch Knospung (englisch budding) freigesetzt werden. Die Virushülle hat eine große Bedeutung bei der Aufnahme von Viren in die Zelle, der Stabilität gegenüber Umwelteinflüssen und Desinfektionsmitteln sowie der erleichterten Fähigkeit zur Veränderung der Virusoberfläche. Diese Variabilität durch eine Virushülle ist ein evolutionärer Vorteil gegenüber unbehüllten Viren. Sie ermöglicht behüllten Viren, die Immunabwehr eines Wirtes leichter zu unterlaufen oder sich besser an einen neuen Wirt anzupassen. Deutlich werden diese Eigenschaften der Virushülle beispielsweise daran, dass sämtliche beim Menschen neu auftretenden Viren (Emerging Viruses), die eine reale oder potentielle Gefährdung durch eine Pandemie darstellen, behüllte Viren sind, so z. B. das HI-Virus, SARS-Coronavirus 1 und 2, Influenzavirus, Ebolavirus und West-Nil-Virus.
Entdeckung
Die Anfänge der Virologie und die Definition der Viren als neue Art infektiöser Erreger sind mit zwei unbehüllten Viren verknüpft: Dem Tabakmosaikvirus (Dmitri Iwanowski 1892 und Martinus Beijerinck 1898) und dem Maul-und-Klauenseuche-Virus (Friedrich Loeffler und Paul Frosch 1897).[1] Das von Walter Reed 1901 entdeckte Gelbfieber-Virus[2] war das erste beim Menschen identifizierte Virus und zugleich das erste beschriebene behüllte Virus. Diese Untersuchungen beschränkten sich jedoch auf Übertragungswege, die Morphologie der Viren blieb bis auf die Eigenschaft der besonderen Kleinheit (Unsichtbarkeit im Lichtmikroskop) zunächst unbekannt.
Diese Barriere der ungenügenden mikroskopischen Auflösung konnte erst in den 1930er-Jahren mit der Entwicklung des Elektronenmikroskops durch Helmut und Ernst Ruska überwunden werden. Schon die ersten Aufnahmen mit dieser neuen Technik zeigten Umrisse von Viren mit länglicher oder runder Gestalt.[3] Eine Differenzierung der Feinstruktur der Viren und Darstellung der Virushülle war mit den frühen Kontrastfärbungen jedoch noch nicht möglich. Immerhin schlug Helmut Ruska 1943 nach Untersuchung damals vorhandener Virusisolate eine erste Einteilung der Viren nach Größe und Form vor.[4] Bis dahin wurden die Viren nach dem befallenen Wirt und der jeweiligen Erkrankung eingeteilt.
In den 1950er-Jahren konnten auch Viren in den von Renato Dulbecco und Harry Eagle entwickelten Zellkulturen gezielt angezüchtet und in großen Mengen vermehrt werden. Durch die Reinheit und Konzentration dieser Viruspräparation wurde die genauere Bestimmung der chemischen Zusammensetzung und damit des Lipidanteils von Viren möglich. Bis zur Etablierung dieser Technik musste man sich auf die Virusisolierung aus infizierten Wirten oder auf die 1932 entwickelte[5] und 1946 für die Virusvermehrung verbesserte Anzucht in bebrüteten Hühnereiern beschränken.[6] Einige Viren verloren ihre Fähigkeit, die Hühnerembryonen zu infizieren, wenn man die Viruslösung vorher mit verschiedenen Stoffen behandelte, darunter auch fettlösende Verbindungen wie Ether (Diethylether) oder Detergenzien wie Natriumdesoxycholat.[7] Diese sogenannte „Ether-Empfindlichkeit“ von Viren wurde nur bei einigen Viren wie den Influenzaviren oder den Herpesviren beobachtet, andere wie das Poliovirus oder das Maul-und-Klauenseuche-Virus waren auch nach einer Behandlung mit Ether noch infektiös. Die Ether-Empfindlichkeit wurde so zu einem weiteren wichtigen Kriterium bei der Einteilung von Viren und konnte in den 1950er-Jahren auch schon mit dem Nachweis von Lipiden bei gereinigten Viren in Verbindung gebracht werden.[8] Ether-empfindliche Viren wiesen einen Lipidanteil von 20–30 % auf.

Dass der Lipidanteil der Viren im Zusammenhang mit einer Membranstruktur stehen könnte, wurde damals bereits vermutet. Die Existenz von lipidhaltigen Biomembranen bei Zellen konnte schon durch die Untersuchungen von Gorter und Grendel 1925[9] bewiesen werden, und es lag nahe, eine ähnliche Struktur bei lipidhaltigen Viren anzunehmen. Entscheidend war der Beweis, dass die Zusammensetzung der Lipidkomponenten der Viren derjenigen der jeweiligen Wirtszellen ähnelte, in denen die Viren angezüchtet wurden.[10] Der erste Hinweis auf eine Virushülle in elektronenmikroskopischen Bildern kann im Nachhinein in einer Untersuchung von Coriell 1950 nachverfolgt werden. Er isolierte Herpes-simplex-Viren aus Herpesbläschen. Dabei beobachtete er eine eigenartige, runde Form der Viren mit einer zentralen Aussparung, die er als „Doughnut-ähnlich“ beschrieb.[11] Heute wird dieses typische Erscheinungsbild der Herpesviren als „Spiegeleiform“ bezeichnet, dies meint ein ikosaedrisches Kapsid im Innern umgeben von einer sehr dicken Virushülle. Erst ab 1959, als ein besonderes Kontrastierungsverfahren mit Uransalzen für die Elektronenmikroskopie entwickelt wurde,[12] stellte sich die Struktur der Viren viel differenzierter dar, so dass auch die Virushülle sichtbar gemacht werden konnte. Noch heute ist diese sogenannte Negativkontrastfärbung die wichtigste Methode zur elektronenmikroskopischen Darstellung von Viren.
Mit der Erforschung der zellulären Membranen in den 1960er- und 1970er-Jahren ging auch eine Erweiterung des Verständnisses der Virushüllen einher. Dies wurde durch verfeinerte Techniken zur Strukturaufklärung der Hüllproteine wie der Röntgenbeugung, der Gefrierbruch-REM[13] und der NMR-Spektroskopie ermöglicht, aber auch dank neuer Überlegungen über die Eigenschaften von Biomembranen wie dem Flüssig-Mosaik-Modell von Singer und Nicholson.[14] In den letzten zwanzig Jahren lieferte besonders die Kryo-Elektronenmikroskopie entscheidende Einblicke in die Feinstruktur der Virushüllen. Mit dieser Technik ist es möglich, die Form und Anordnung einzelner Hüllproteine zu bestimmen und mit einer fourier-gestützten Bildverarbeitung die Virushülle mit einer Auflösung von 0,6–1 nm darzustellen.
Aufbau der Virushülle
Eine Virushülle besteht immer aus viralen Hüllproteinen, die in eine Phospholipid-Doppelschicht eingebettet sind. Die Einlagerung der Hüllproteine in die Membran geschieht bereits während ihrer Synthese an den Ribosomen des rauen Endoplasmatischen Retikulums (rER). Entweder kann die Virushülle sich bereits hier aus der Membran des rER bilden oder die mit Hüllproteinen besetzten Membranbereiche werden durch den normalen zellulären Membranfluss zur Zellmembran, Kernmembran oder dem Golgi-Apparat transportiert. Dadurch, dass sich die Hüllproteine beim Prozess der Umhüllung in kleineren Membranflächen konzentrieren und zusammenlagern, werden zelluläre Membranproteine verdrängt, die dann nicht in die Virushülle eingebaut werden. Aufgrund dieser Verdrängung zellulärer Membranproteine besteht die Lipid-Doppelschicht der Virushülle nicht aus unveränderten zellulären Membranen, sondern nur aus deren Lipidanteil.
Der Anteil an eingelagerten Hüllproteinen ist meist so hoch, dass der Lipidanteil an der Oberfläche an keiner Stelle unbedeckt vorliegt. Die Lipidmembran der Virushülle ist daher für Antikörper nicht mehr direkt zugänglich. Bei einigen Viren wie beispielsweise den Hepadnaviridae ist der Proteinanteil der Virushülle so hoch, dass die Virushülle fast ausschließlich aus dicht gepackten Hüllproteinen besteht. Diese sind sehr regelmäßig angeordnet und gegenüber Umwelteinflüssen und Detergenzien resistenter als andere behüllte Viren.
Lipidanteil

Die Lipidmembran der Virushülle besteht, wie auch alle zellulären Membranen, aus einer Doppelschicht von Phospholipiden. Diese besitzen einen hydrophilen Kopf, der die Oberflächen der Membran bildet, und zwei nach innen gerichtete, lipophile Kohlenwasserstoffketten. Die am Aufbau der Virushülle beteiligten Phospholipide sind Phosphatidylcholine (auch Lecithine genannt), Phosphatidylethanolamine, Phosphatidylserine, Phosphatidylinositol und Sphingomyeline. Letztere sind nur in der äußeren Schicht der Phospholipidmembran vorhanden. Zu den Phospholipiden tritt noch ein unterschiedlich hoher Anteil an Cholesterin hinzu.[15] Die zellulären Membranen, und damit auch die Virushüllen, variieren in der Zusammensetzung der verschiedenen Phospholipide und dem Gehalt an Cholesterin. Ein hoher Cholesteringehalt ist für die Zellmembran typisch, während die Membranen des Endoplasmatischen Retikulums und des Golgi-Apparates nur wenig Cholesterin enthalten. Der Cholesteringehalt einer Membran, ausgedrückt als C/P-Quotient (Molarer Cholesterin/Phospholipid-Quotient), beeinflusst entscheidend die Morphologie einer Membran, so sind cholesterinreiche Membranen (also mit einem typischen C/P-Quotienten von 0,4 bis 0,8) stabiler, weniger flexibel und mit 5–6 nm um etwa ein Drittel dicker als cholesterinarme.[16] Da die Lipidzusammensetzung einer Virushülle in erster Näherung jener der ursprünglichen zellulären Membran entspricht, sind diese Unterschiede auch zwischen Virushüllen zu finden, die von der Zellmembran oder von intrazellulären Membransystemen abstammen.
Bei genauer Betrachtung weicht die Lipidzusammensetzung der meisten Virushüllen in geringem Umfang von ihrer Ursprungsmembran ab. Wie diese selektive Aufnahme von Lipidkomponenten in die Virushülle geschieht, ist derzeit noch unklar. Man vermutet einen bevorzugten Einbau von verschiedenen Phospholipiden während der Aggregation von Hüllproteinen in der Membran, wobei die Hüllproteine mit den Lipiden unterschiedlich stark wechselwirken und die Bindung der Hüllproteine untereinander bestimmte Phospholipide bevorzugt. Die Entdeckung der sogenannten Lipid Rafts, also in einer zellulären Membran schwimmenden Mikroareale mit hohem Cholesteringehalt, hat einen inhomogenen Aufbau dieser Membranen aufgezeigt. Diese Lipid Rafts scheinen auch für den selektiven Einbau in die Virushülle bedeutsam zu sein. Bei einigen Viren ist die Präsenz dieser Mikroareale sogar für die Einlagerung der Hüllproteine und die Entstehung der Virushülle eine notwendige Voraussetzung,[17] da sie die Dichte viraler Hüllproteine regional erhöhen und damit die Aggregation ermöglichen. Umgekehrt ist die Präsenz von Cholesterin in der Virushülle einiger Viren für das Eindringen in die Zelle notwendig. So verminderte die Anzüchtung des Caninen Staupevirus in Zellkulturen, denen ein Hemmstoff für die Cholesterinsynthese beigegeben wurde, seine Fähigkeit weitere Zellen zu infizieren um 80 %.[18] Gleiches wurde auch bei Virushüllen des Varizella-Zoster-Virus beobachtet, die nicht an der cholesterinreichen Zellmembran entstehen.[19]
Tabelle: Vergleich der Lipidkomponenten von typischen zellulären Membranen (rER: raues ER, sER: glattes ER) und Virushüllen (Humanes Immundefizienzvirus HIV-1, sphärische Antigen-Partikel des Hepatitis-B-Virus sHBV). Zur besseren Vergleichbarkeit sind die Lipidkomponenten jeweils in molare Prozent des Lipidanteils für die Leberzelle der Ratte aus den Daten von M. K. Jain (1980)[20] umgerechnet. Die sER-Membran enthält zusätzlich 2,0 % Diphosphatidylglycerin (Cardiolipin).
| Lipidkomponente | Zellmembran | rER-Membran | sER-Membran | Golgi-Membran | HIV-1[21] | sHBV[22] |
|---|---|---|---|---|---|---|
| Cholesterin | 34,5 | 6,6 | 10,4 | 9,1 | 46,8 | 3,1 |
| Phosphatidylcholin | 20,7 | 60,4 | 56,9 | 48,8 | 12,7 | 78,9 |
| Sphingomyelin | 16,0 | 3,3 | 12,4 | 12,2 | 15,1 | 1,9 |
| Phosphatidylethanolamin | 12,6 | 17,6 | 21,7 | 18,3 | 13,1 | 9,2 |
| Phosphatidylinositol | 4,6 | 8,8 | 6,9 | 7,3 | 1,1 | 3,6 |
| Phosphatidylserin | 10,3 | 3,3 | — | 4,3 | 8,0 | 0,9 |
| Lipidgehalt % | 87,0 | 91,0 | 96,6 | 82,0 | 28,0 | 24,0 |
| C/P-Quotient | 0,53 | 0,07 | 0,11 | 0,10 | 0,88 | 0,03 |
Wie aus einem Vergleich der in obiger Tabelle aufgelisteten Lipidkomponenten hervorgeht, kann man durch die Bestimmung der Lipidzusammensetzung einer Virushülle auf die zelluläre Ursprungsmembran rückschließen. Im angeführten Beispiel besitzt das HIV-1 die typische Zusammensetzung der Zellmembran und die sphärischen, leeren HBV-Partikel − die in ihrer Lipidzusammensetzung den kompletten Virionen entsprechen − dem Lipidprofil des rauen ER. Die spezifische An- und Abreicherung der Komponenten im Vergleich zur Ursprungsmembran ist ebenfalls erkennbar. Da die Lipidzusammensetzung der Membranen verschiedener Zelltypen variieren kann, sind auch entsprechende Abweichungen der Virushülle bei einem Virus zu erwarten, wenn es sich im Organismus oder in der Zellkultur in verschiedenen Zelltypen vermehrt. Auch kann es innerhalb einer einzigen Zelle zu leicht unterschiedlichen Lipidanteilen kommen, wenn die Zellmembran eine gerichtete Polarität besitzt wie z. B. bei Zellen, die an einem Lumen angeordnet sind. So können sich an apikalen oder basalen Bereichen der Zellmembran unterschiedliche Virushüllen bilden.[23]
Die Bedeutung der Lipidkomponenten für die wichtigen Funktionen der Virushülle wie Virusaufnahme, Infektiosität, Membranfusion und Zusammenbau der Viruspartikel wurde lange Zeit nicht erkannt, da das Hauptaugenmerk auf der Erforschung der Hüllproteine lag. Viele Untersuchungen an unterschiedlichen Viren zeigen jedoch in jüngster Zeit, wie sehr die Lipide durch Protein-Lipid-Interaktionen die Funktion der Hüllproteine erst ermöglichen; insbesondere die Anreicherung von Cholesterin in den Lipid Rafts scheint die Funktion der Virushülle entscheidend zu beeinflussen.[24] Die Lipidmembran hat hierbei einen erheblichen Einfluss auf die Anordnung der Hüllproteine und ihre korrekte Faltung als Tertiärstruktur.
Virale Hüllproteine
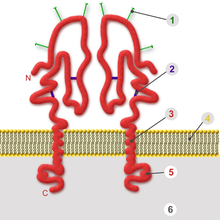
In ähnlicher Weise wie zelluläre Transmembranproteine sind die viralen Hüllproteine (englisch envelope proteins; im Unterschied zu Kapsidproteinen, en. coat protein, capsid protein, CP) in die Lipidmembran eingelagert. Eine oder mehrere transmembranäre, lipophile Proteindomänen durchqueren die Lipidmembran und trennen damit eine kleinere innere Domäne von einer größeren äußeren. Bei den meisten Hüllproteinen liegt der Carboxyl-Terminus innen, so dass die Hüllproteine zu den Klasse-1-Membranproteinen gehören.
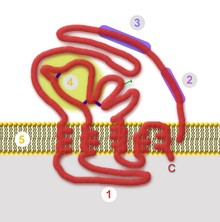
Die nach innen gerichtete Domäne (auch „intrazellulärer Anker“ oder Ankerdomäne genannt) ist hydrophil und kann die Bindung an nachfolgende innere Strukturen vermitteln. Im klassischen Fall ist dies ein Kapsid. Bei Viren mit mehreren Kapsiden oder komplex aufgebauten Viren bindet die innere Domäne an weitere Proteine, die die Unterseite der Virushülle zusätzlich auskleiden. Diese liegen zwischen Kapsid und Hülle im Matrixraum und werden daher als Matrixproteine bezeichnet. Im einfachsten Falle besteht die innere Domäne des Hüllproteins aus einem gefalteten Ende des Proteins. Durchquert das Hüllprotein mehrmals die Lipidmembran („multipass“), ist die innere Domäne eine sich daraus ergebende Schleife. Die Wechselwirkung zwischen den inneren Domänen, entweder direkt ohne weitere Bindungspartner oder indirekt über Matrixproteine oder Kapsid, ist die bestimmende Kraft zur Krümmung der Membran während der Umhüllung.
Die Transmembran-Domäne besteht aus einer lipophilen α-Helix, deren Länge der Dicke der Lipidmembran angepasst ist. Jene Viren, die an der dickeren, cholesterinreichen Zellmembran umhüllt werden, benötigen für die Helix z. B. 26 Aminosäuren (Influenzavirus). Werden die Viren an der Membran des rER umhüllt, genügen 18–20 Aminosäuren (Gelbfieber-Virus) für eine transmembranäre Helix. Die Strukturaufklärung eines Hüllproteins mag daher einen Hinweis darauf geben, an welcher Membran die Virionen gebildet werden. Das virale Hüllprotein kann auch mehrere Transmembran-Domänen besitzen, deren Helices eng aneinander liegende Bündel in der Membran bilden. Die Hüllproteine der Familie Flaviviridae besitzen zwei transmembranäre Helices, deren enge Bindung aneinander durch eine hydrophile Flanke vermittelt wird; diese Domänen besitzen somit eine amphiphile Struktur.[25] Da die Helices die Lipide der Membran verdrängen, kann man die allgemeine Regel aufstellen, dass der Lipidanteil einer Virushülle umso geringer wird, je mehr transmembranäre Domänen die Hüllproteine besitzen.
Der äußere Teil eines Hüllproteins ist meist an vielen Stellen glykosyliert, also mit kurzen Zuckerresten (Oligosaccharide) kovalent verknüpft, weshalb virale Hüllproteine zu den Glykoproteinen gezählt werden. Dieser äußere Teil des Hüllproteins ist wesentlich für die Bindung an Rezeptoren und die Membranfusion bei der Virusaufnahme. Die äußeren Domänen werden auch durch Antikörper der Immunabwehr erkannt, so dass sich in exponiert gelegenen Epitopen oft sehr variable Abschnitte befinden, die man meist als hypervariable Regionen (HVR) bezeichnet. Die HVR der Hüllproteine führen zu einer hohen immunologischen Flexibilität des Virus, da sie durch häufige Mutationen die Bindung von Antikörpern einschränken und sich an unterschiedliche Zellrezeptoren neuer Wirte schnell anpassen können.
Die Aufgaben der äußeren Domäne – Rezeptorbindung und Membranfusion – können in einem Hüllprotein vereinigt oder auf mehrere, kooperierende Hüllproteine verteilt sein. Mit nur wenigen Ausnahmen lagern sich die Hüllproteine zu Komplexen aus mehreren gleichen oder verschiedenen Hüllproteinen zusammen. Diese Oligomere können bei entsprechender Größe in der elektronenmikroskopischen Darstellung als sogenannte „Spikes“ oder Peplomere sichtbar werden. Sehr charakteristische Spikes lassen sich beispielsweise bei den Virusfamilien Orthomyxoviridae und Coronaviridae darstellen; letztere erhielten durch diese Charakteristik der Virushülle auch ihren Namen.
Die Anzahl verschiedener Hüllproteine und die Zusammensetzung der Hüllprotein-Oligomere ist für viele Virusgattungen charakteristisch. Nur ein Hüllprotein liegt bei den Rhabdoviren vor, das einfache Trimere bildet ([G]3). Bei Retroviren, z. B. dem Rous-Sarkom-Virus, lagern sich zwei Glykoproteine (SU und TM) zu einem Heterodimer zusammen, das sich wiederum mit zwei weiteren Heterodimeren zu einem Hexamer anordnet ([SU-TM]3). Alphaviren verfügen über zwei (E1, E2) oder drei Hüllproteine (E1-3), die sich nach Zusammenlagerung zu größeren Dreierkomplexen anordnen ([E1-E2-E3]3).
Bei Viren, die an der Zellmembran knospen, werden die Hüllproteine zunächst in die Zellmembran eingelagert und diese damit mit Proteinen angereichert, welche die Fähigkeit zur Membranfusion besitzen. Diese Veränderung der Zellmembran kann dazu führen, dass Zellmembranen benachbarter Zellen durch die viralen Hüllproteine miteinander fusionieren können und damit Riesenzellen, sogenannte Synzytien bilden. Dies kann für das Virus von Vorteil sein, da sich mit der Verschmelzung von Zellen die Infektion ausbreiten kann und ein größerer Syntheseapparat für die Viren zur Verfügung steht; dies ist z. B. beim Respiratory-Syncytial-Virus der Fall. Die Fusion von Zellen in der Haut durch Hüllproteine des Masernvirus[26] verursacht eine lokale Entzündung, die dann als typische rötliche Flecken einer Maserninfektion sichtbar werden. Viele Viren haben jedoch Strategien entwickelt, um die Bildung von Synzytien zu verhindern, da diese auch das Knospen neuer Viren behindern kann. Die Fusionseigenschaft der Hüllproteine wird in diesem Fall erst durch einen zusätzlichen Reifungsschritt aktiviert, in dem die Fusionssequenz entweder erst nach einem Verdau durch eine Protease oder Glykosidase freigelegt wird oder ein saures Milieu (pH < 6,0) eine Konformationsänderung des Hüllproteins herbeiführt, durch die die Fusionssequenz erst nach außen gestülpt wird. Das bekannteste Beispiel ist das Hüllprotein Hämagglutinin der Orthomyxoviren, das erst durch eine Neuraminidase aktiviert werden muss und eine pH-abhängige Fusionsaktivität besitzt.
Hüllproteine erfüllen in der Zellmembran auch gelegentlich andere Funktionen während der Virusvermehrung als nur die Umhüllung des Virions. Hierzu können sie sich alternativ zu neuen Strukturen anordnen und wie beispielsweise beim SARS-Coronavirus Poren bilden, die zur Lyse der Zelle führen.[27]
Symmetrische Virushüllen
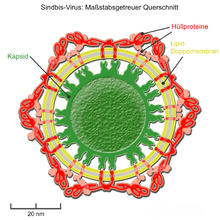
Der innere Anteil der Hüllproteine kann mit einem umhüllten Kapsid dergestalt interagieren, dass stets nur ein Hüllprotein (oder ein zusammengelagertes Dimer bzw. Trimer der Hüllproteine) an nur ein Kapsomer bindet. Durch diese feste Anordnung wird die Form und Symmetrie des inneren ikosaedrischen Kapsids auf die äußere Virushülle übertragen und es ergeben sich trotz der Beweglichkeit der Lipidmembran streng ikosaedrisch aufgebaute Virushüllen.[28] Diese Form der sogenannten „Morphogenese von innen nach außen“ findet sich bei der Gattung Alphavirus der Familie Togaviridae (z. B. dem Semliki-Forest-Virus[29] und Sindbis-Virus) und der Gattung Flavivirus der Familie Flaviviridae.
Bei den größeren Viren der Familie Bunyaviridae (80–120 nm) ist ebenfalls eine regelmäßige Anordnung der Hüllproteine in Form eines Ikosaeders nachweisbar (Triangulationszahl T=12), jedoch kein symmetrisches, ikosaedrisches Kapsid, das diese Symmetrie von innen stützen könnte.[30] Hier gibt die enge Wechselwirkung der Hüllproteine untereinander die Form der Virushülle vor, was man auch als eine „Morphogenese von außen nach innen“ bezeichnen kann. Bei den Bunyaviren werden drei helikale Kapside in der Virushülle verpackt, was ähnlich den Influenzaviren einen Austausch der RNA-Segmente (Reassortment) und eine hohe genetische Flexibilität ermöglicht. Diese Flexibilität der Influenzaviren aufgrund mehrerer, unregelmäßiger Kapside bzw. RNA-Stränge wird durch eine ungeordnete und damit sehr instabile Virushülle erkauft, die bereits durch Austrocknung und milde Detergenzien inaktiviert werden kann. Der Familie Bunyaviridae hingegen verleiht die symmetrische und somit festere Anordnung der Hüllproteine eine vergleichsweise hohe Stabilität, so dass diese − wie z. B. bei der Gattung Hantavirus − in ausgetrocknetem Zustand monatelang infektiös bleiben und selbst eine Ausscheidung über den Urin überstehen, obwohl Harnstoff als Detergens wirkt und andere behüllte Viren inaktiviert.
Sonderformen
Bei wenigen Virusfamilien ist eine Phospholipidmembran nicht als äußere, umhüllende Struktur vorhanden, sondern befindet sich im Inneren der Virionen. Besonders außergewöhnlich sind hier zwei Familien von Bakteriophagen, die Corticoviridae und Tectiviridae, bei denen sich die Lipidmembran im Inneren eines ikosaedrischen Kapsids befindet. Diese Struktur wird nicht als Virushülle bezeichnet, da sie weder außen liegt noch typische Aufgaben einer Virushülle wie die Anheftung an die Zelloberfläche erfüllt. Das bei den Tectiviridae vorhandene Membranbläschen dient nach Anheftung des Kapsids an die Bakterienoberfläche dem aktiven Eindringen der doppelsträngigen Bakteriophagen-DNA in die Wirtszelle.
Bei Vertretern der Familie Poxviridae besteht die Virushülle aus einer Doppelmembran mit einer äußeren und zusätzlich inneren Phospholipid-Doppelschicht. Innerhalb des Zytosols liegen die Pockenviren mit einer einfachen Umhüllung vor. Diese erste Umhüllung entsteht nicht durch Knospung aus einer zellulären Membran, sondern durch den Zusammenbau einer vollständig neuen Lipidmembran an der Außenseite des noch unreifen, später doppelkonkaven Kapsids. Zum Neuaufbau der Membran werden abgebaute Membranbestandteile aus dem Übergangsbereich zwischen Golgi- und ER-Membran (Intermediäres Kompartiment) verwendet.[31] Das einfach umhüllte Viruspartikel erhält dann durch Knospung an der Golgi-Membran eine zweite, äußere Virushülle.
Entstehung während der Virusvermehrung
Die Synthese der Hüllproteine und die Entstehung der Virushülle markiert die letzte Phase im Vermehrungszyklus eines behüllten Virus; zuvor muss das virale Genom repliziert und eventuell in ein Kapsid verpackt werden.[32]
Der Vorgang der Umhüllung eines Virus, auch Knospung („budding“) genannt, entspricht einer spezifischen Verpackung in einem abgeschnürten Membranbläschen. Innerhalb von Zellen ist die ständige Bildung und Fusion von Membranbläschen ein physiologischer Vorgang zum Stofftransport, der sogenannten Exozytose bzw. Endozytose. Ein behülltes Virus nutzt diese schon vorhandenen Eigenschaften und Mechanismen des Membranflusses, in dem es diesen modifiziert und durch die viralen Strukturproteine steuert. Die Energie, die zur Krümmung der Lipidmembran und zur Bläschenbildung erforderlich ist, entstammt ausschließlich der Wechselwirkung der Hüllproteine untereinander, der Hüllproteine mit inneren Strukturen wie Matrixproteinen und Kapsiden oder der Kapside mit der Lipidmembran; eine Zufuhr von Energie beispielsweise in Form von ATP ist hierzu nicht notwendig. Die energetisch günstigere Zusammenlagerung der Hüllproteine überwindet beispielsweise bei Togaviren die für die Lipidmembran energetisch ungünstigere Krümmung mittels Wasserstoffbrückenbindungen, ionischen Bindungen und besonders durch hydrophobe Wechselwirkungen. Die Entstehung der Virushülle – gleichgültig an welchem Membransystem – wird daher lediglich von der Translation, dem Transport und der Konzentration der Virusproteine am jeweiligen Membrankompartiment gesteuert. Die Knospung als spontane Zusammenlagerung von Kapsid, Lipidmembran und Hüllproteinen, ist im Hinblick auf ihre thermodynamische Betrachtung von großem Interesse. Zu ihrer Beschreibung wurden vielfach Modelle herangezogen, um die Wechselwirkung der beteiligten Komponenten berechnen zu können.[33] In Übereinstimmung mit der in vitro gemessenen Dauer, die der Knospungsvorgang in Anspruch nimmt, konnten auch im Modell 10 bis 20 Minuten berechnet werden.[34] Als limitierende Prozesse wurden die Diffusion der Hüllproteine entlang der Lipidmembran und die Verdrängung von Wassermolekülen zwischen Hülle und Kapsid abgeleitet. Die Modellrechnungen lassen auch eine bevorzugte Knospung von Viren an jenen Stellen der zellulären Membranen erwarten, an denen bereits morphologisch eine Krümmung der Lipidmembran vorliegt. Dies stimmt mit den elektronenmikroskopischen Untersuchungen von infizierten Zellen überein, in denen knospende Viren überwiegend an den gekrümmten Seiten des Golgi-Apparates oder der Zellmembran gefunden wurden.
Die Mechanismen der Umhüllung sind an der jeweiligen zellulären Membran im Prinzip ähnlich. Das erste Modell zur Knospung wurde am Beispiel des Semliki-Forest-Virus (SFV) entwickelt. Hier führt die Bindung des intrazellulären Ankers der Hüllproteine an ein bereits geschlossenes Kapsid zur Krümmung der Lipidmembran.[35] Daraus wurde die These abgeleitet, dass zur Entstehung einer Virushülle das Vorhandensein von Hüllproteinen und die Bindung an Kapsidproteine zwingend notwendig sei. Dieses frühe Modell wurde erheblich eingeschränkt, als man bei Retroviren eine Umhüllung von Kapsiden auch ohne Anwesenheit der Hüllproteine (Env-Protein) beobachtete, wenn die Kapsidproteine (Gag-Proteine) alleine in transfizierten Zellkulturen zur Verfügung stehen.[36]
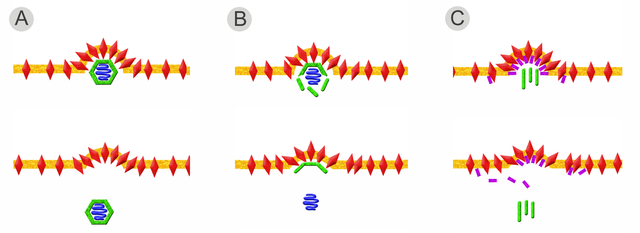
Neben dem Spezialfall für das Gag-Protein der Retroviren, gibt es drei weitere, wichtige Varianten der Knospung (vgl. Abbildung). Der einfachste Weg ist die leichte Krümmung der Lipidmembran durch die Wechselwirkung der Hüllproteine über ihre inneren Ankerdomänen. Ein geschlossenes Kapsid bindet an diese aggregierten Hüllproteine und treibt durch die Interaktion mit den Hüllproteinen die Knospung voran (siehe Abbildung, Fall A). Im zweiten Fall (B) erfolgt der Zusammenbau des Kapsids erst nach Bindung an die Hüllproteine. Die Interaktion von Kapsidproteinen und Hüllproteinen ermöglicht erst die Bindung der Nukleinsäure und komplettiert das Virus während der Knospung. Diese Variante kann auch durch ein zwischen Hülle und Kapsid vermittelndes Matrixprotein ergänzt werden. Bei Viren, die keine symmetrischen Kapside besitzen (beispielsweise das Bovine Virusdiarrhoe-Virus und das verwandte Hepatitis-C-Virus), genügt die Bindung der Nukleinsäure an basische Proteine (Nukleo- oder Coreproteine), die ähnlich den Matrixproteinen an der Innenseite der Membran mit den Hüllproteinen interagieren.
Bei der dritten Variante (C) wird die Interaktion der intrazellulären Anker der Hüllproteine erst durch die Bindung an Matrixproteine ermöglicht. Nachdem diese Interaktion zu einer ersten Krümmung der Lipidmembran geführt hat, kann nun ein Kapsid (wie bei den Herpesviren) oder auch mehrere helikale Kapside (wie bei den Orthomyxoviridae) gebunden und umhüllt werden.
- Entstehung der Virushülle durch Knospung: TEM-Abbildungen von infizierten Zellkulturen
-
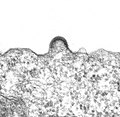 Aggregation von Hüllproteinen und Krümmung der Zellmembran bei HIV-1
Aggregation von Hüllproteinen und Krümmung der Zellmembran bei HIV-1 -
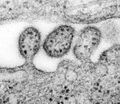 Lassa-Virus in der späten Phase der Abschnürung der Virushülle
Lassa-Virus in der späten Phase der Abschnürung der Virushülle -
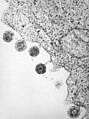 Humanes Herpesvirus 6 nach Freisetzung an der Zellmembran
Humanes Herpesvirus 6 nach Freisetzung an der Zellmembran -
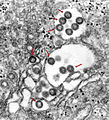 Behüllte Partikel des Rifttalfieber-Virus (Bunyaviridae) im Lumen des ER
Behüllte Partikel des Rifttalfieber-Virus (Bunyaviridae) im Lumen des ER
Knospung an der Zellmembran

Die Entstehung der Virushülle an der Zellmembran erfordert zunächst einen Transport der Hüllproteine an die Zelloberfläche. Die viralen Proteine entstehen an den Ribosomen des rauen ER, wobei die Hüllproteine noch während der Synthese mit ihrer transmembranären Domäne die Membran des ER durchstoßen und in sie eingelagert werden. Über das Membransystem des Golgi-Apparates werden die Hüllproteine glykosyliert. Die nun modifizierten (reifen) Hüllproteine werden in abgeschnürten, exozytotischen Vesikeln an die Zellmembran transportiert und fusionieren mit ihr. Jene Domänen der Hüllproteine, die zuvor in das Lumen des ER gerichtet waren, sind nun extrazellulär angeordnet. Die an die Zellmembran herangeführten restlichen Viruskomponenten (Kapside, Nukleinsäure und eventuelle Matrixproteine), können nun umhüllt werden.[37] Der Entstehungsweg über die Zellmembran setzt die Einlagerung viraler Hüllproteine voraus, was zu der bereits erwähnten Bildung von Synzytien führen kann (siehe Abschnitt Hüllproteine). Diese nach außen präsentierten Virusproteine können jedoch zusätzlich von Immunzellen als fremd erkannt werden, so dass eine frühe Immunantwort gegen die Hüllproteine erfolgen kann. Alle Viren, deren Hüllen sich von der Zellmembran ableiten, werden durch Fusion der Hülle mit der Zellmembran auch wieder aufgenommen. Diese Art der Aufnahme (fusion from without) ermöglicht eine Infektion ohne einen Transport in einem Endosom.
Wichtige Virusfamilien, die sich durch eine Knospung an der Zellmembran auszeichnen, sind beispielsweise die Retroviridae, Orthomyxoviridae, Togaviridae und alle Virusfamilien mit einer einzelsträngigen RNA negativer Polarität (ss(-)RNA) als Genom (Ordnung Mononegavirales), also den Bornaviridae, Rhabdoviridae, Filoviridae und Paramyxoviridae.
Knospung an der Golgi- und ER-Membran
Da die Hüllproteine zunächst immer in die Membranen der intrazellulären Membransysteme eingelagert sind, kann auch schon hier eine Knospung erfolgen. Bei diesem Entstehungsweg kann entweder die Lipidmembran des ER oder – nach Vesikeltransport – des Golgi-Apparates gewählt werden. Dies wird vorwiegend durch die eventuell notwendigen Modifikationen der Hüllproteine bestimmt, die fast nur durch die Enzyme des Golgi-Apparates vollzogen werden können. Sehr häufig erfolgt die Knospung am Übergangsbereich der beiden Membransysteme, dem sogenannten Intermediären Kompartiment. Das behüllte Viruspartikel befindet sich nach der Knospung stets im Lumen der Membransysteme, von wo sie im Inneren eines Transportvesikels (Exosom) nach außen befördert werden. Diese intrazelluläre Behüllung der Viren kann sich ohne Zerstörung der Zelle vollziehen, da keine Virusproteine die Zelloberfläche verändern und zur Ausschleusung der Viren die normale Exozytose genutzt wird. Bei der Infektion der nächsten Wirtszelle müssen alle diese Viren von einem Endosom aufgenommen werden, mit dessen Membran die Virushülle fusioniert (fusion from within). Wichtige Virusfamilien mit einer intrazellulären Knospung sind die Coronaviridae, Hepadnaviridae und Flaviviridae.
Knospung an der Kernmembran
Die Mitglieder der Virusfamilie Herpesviridae sind in ihrem Aufbau, ihrer Vermehrungsstrategie und auch in der Entstehung der Virushülle ein Sonderfall, da die sehr großen Kapside der Herpesviren im Zellkern zusammengebaut werden, in dem auch die doppelsträngige DNA der Viren synthetisiert wird. Bereits bei sehr frühen elektronenmikroskopischen Untersuchungen an Zellen, in denen sich das Herpes-simplex-Virus vermehrt, konnte man knospende Kapside an der Innenseite der Kernmembran und behüllte Viruspartikel in der den Kern umgebenden perinukleären Zisterne erkennen.[38] Da die perinukleäre Zisterne über Membranschläuche mit dem rauen ER verbunden ist, nahm man an, dass die umhüllten Virionen dann über Membranbläschen des Golgi-Apparates aus der Zelle geschleust werden. Eine Untersuchung der Lipidzusammensetzung der Virushülle ergab jedoch, dass die Lipidkomponenten nicht denen der Kernmembran entsprechen, sondern das Lipidprofil der Golgimembran besitzen.[39] Dieser Befund führte zu der Entdeckung, dass die Herpesviren zuerst durch Knospung an der Kernmembran eine Virushülle erlangen. Diese fusioniert jedoch wieder mit der äußeren Membran der perinukleären Zisterne und gibt so das nackte Kapsid in das Zytosol frei. Erst durch eine zweite Knospung in ein abgeschnürtes Membranbläschen des Golgi-Apparates, das mit viralen Hüll- und Matrixproteinen angereichert ist, erhält das Kapsid seine endgültige Hülle. Diese sogenannte sekundäre Behüllung entspricht dann erst der Virushülle der freigesetzten Viren.
Leere Virushüllen und „Defekte Viren“
Bei einigen Viren sind die Hüllproteine in der Lage, ohne eine weitere Bindung an eine innere Struktur eine Knospung hervorzurufen. Dies ist insbesondere dann der Fall, wenn die Wechselwirkung zwischen den inneren Ankerdomänen der Hüllproteine besonders hoch ist. Das Resultat sind leere oder unvollständig gefüllte Virushüllen. Die Existenz dieser leeren Hüllen wurde zuerst bei Untersuchungen des sogenannten „Australia Antigen“ entdeckt, das zur Entdeckung des Hepatitis-B-Virus (HBV) durch B. Blumberg führte. Das entdeckte Antigen besteht aus den drei Hüllproteinen des HBV (HBs-Antigen). Im Blut von HBV-Infizierten ist das HBs-Antigen überwiegend in leeren, sphärischen Partikeln mit einem Durchmesser von 22–24 nm und leeren schlauchartigen Strukturen („Tubuli“) von variabler Länge zu finden.[40] Unter etwa 1.000 bis 10.000 HBs-Antigen-haltigen Partikeln ist nur ein infektiöses, komplettes Virus (42 nm) nachzuweisen. Dieser gewaltige Überschuss an leeren Virushüllen dient vorrangig dazu, Antikörper gegen das Hüllprotein zu neutralisieren und damit ihre Bindung an die kompletten Viren zu verhindern.
Leere Virushüllen, die wie im Beispiel des HBV oft kleiner sind als die kompletten Viren, werden auch bei einer fehlerhaften oder unvollständigen Verpackung segmentierter Genome (z. B. Influenzavirus) gefunden, wenn sie in Zellkulturen angezüchtet werden. Diese Partikel werden auch als defective interfering particles (DIP) oder virus-like particles (VLP) bezeichnet. Beim Hepatitis-C-Virus wurde die Existenz unvollständiger Partikel im Blutserum von Patienten vermutet, da ein wechselndes stöchiometrisches Verhältnis von Coreprotein zu RNA nachweisbar ist.[41]
Ein besonderes Beispiel der Virushülle liefert das Hepatitis-D-Virus, da es selbst keine Gene für eine ausreichende Verpackung mit Hüllproteinen besitzt. Es ist auf die Anwesenheit des HBV in derselben Zelle angewiesen, da es nur mit den Hüllproteinen des HBV verpackt und freigesetzt werden kann. Es wird daher als defektes oder abhängiges Virus (Virusoid) bezeichnet.[42]
Die Kapside behüllter und unbehüllter Viren
Bei Virushüllen mit hohem Lipidanteil sind die Hüllproteine flexibel angeordnet und können sich seitwärts in der Membran bewegen. Diese flüssige Eigenschaft der Virushülle bedeutet, dass auch dann eine geschlossene Umhüllung vorliegt, wenn ein Fehler in der Anordnung der Hüllproteine oder eine Lücke in der Oberflächensymmetrie auftritt. Eine solche Fehlanordnung würde bei unbehüllten Viren zu einem mangelhaften Schutz des Genoms oder zum Zerfall des Kapsids führen. Unter dem Schutz einer Virushülle besteht für die Struktur des Kapsids im Vergleich zu unbehüllten Viren eine größere Freiheit, da diese nicht mehr unmittelbar dem Schutz des Genoms vor Nukleasen dienen oder einen Angriffspunkt für das Immunsystem darstellen. Die Kapside behüllter Viren können daher auch Lücken aufweisen oder nur netzartig das Genom umkleiden. Dies hat bei Retroviren und den nahe verwandten Hepadnaviren (z. B. dem Hepatitis-B-Virus)[43] eine große Bedeutung, da das noch unbehüllte, aber geschlossene Kapsid während der Vermehrung noch ATP und Nukleotide aufnehmen kann, um das schon verpackte Genom zu komplettieren. Bei den Kapsiden einiger behüllter Viren lassen die Lücken auch eine Freisetzung des Genoms z. B. an der Kernpore zu, ohne dass das Kapsid im Cytosol vorher zerfallen muss.
Biologische Bedeutung
Virushülle als Pathogenitätsfaktor
Als äußere Struktur ist die Virushülle für alle Eigenschaften eines Virions verantwortlich, die den Infektionsweg, die Aufnahme in die Zelle und die Abwehr durch das Immunsystem betreffen. In dieser durch die Virushülle vermittelten Auseinandersetzung mit dem Wirtsorganismus haben sich im Laufe der viralen Evolution Mechanismen herausgebildet, die für die Vermehrung des Virus von Vorteil sind und als Virulenz- oder Pathogenitätsfaktoren bezeichnet werden. Eines dieser Phänomene ist das sogenannte Molekulare Mimikry, also die Nachahmung von Proteinen des Wirtsorganismus durch Hüllproteine, die dadurch vom Immunsystem nicht mehr als fremd erkannt werden oder sogar Funktionen dieser Proteine nachahmen können.
Ein Beispiel für diese immunologische Tarnung ist die Ähnlichkeit von Teilen des Hüllproteins einiger Virusarten der Familie Coronaviridae mit dem Fc-Fragment des IgG-Antikörpers.[44] Auch das Hüllprotein E2 des Hepatitis-C-Virus ist strukturell einem Teil des IgG-Antikörpers (ab-Fragment) ähnlich.[45] Neben einer solchen Tarnung durch strukturelle Nachahmung kann auch die spezifische Bindung von Wirtsproteinen an die Virushülle genutzt werden. Dies liegt im Falle der spezifischen Bindung von Albumin an die Hülle des Hepatitis-B-Virus vor.[46]
Neben der reinen Nachahmung wirtseigener Proteine zur Tarnung können die Hüllproteine auch Bindungseigenschaften der Wirtsproteine imitieren. Bei den Retroviren der Gattung Lentivirus ist die Ähnlichkeit der äußeren Domäne des Hüllproteins gp41 mit Interleukin-2 beschrieben worden; hier wird die Bindung an Interleukin-Rezeptoren von Immunzellen imitiert, die als Zielzellen dieser Viren gelten.[47]
Die Fähigkeit von Retroviren in der Zellkultur, ohne ein eigenes Hüllprotein eine Knospung zu induzieren, wird bei der gentechnischen Erzeugung von künstlichen Viruspartikeln genutzt, um Partikel mit veränderten Oberflächeneigenschaften herzustellen. So können in die Hülle dieser sogenannten Pseudotypen fremde Hüllproteine eingelagert werden, um beispielsweise die Bindung dieser an Rezeptoren untersuchen zu können oder sie in der Forschung als virale Vektoren einzusetzen. Die Bildung von Pseudotypen scheint an die Existenz der schon erwähnten Lipid Rafts gebunden zu sein.[48]
Auch bei natürlichen Infektionen ist die Entstehung von Pseudotypen beschrieben. So können zwei Virusarten bei gleichzeitiger Infektion einer Zelle die unterschiedlichen Hüllproteine gemischt in eine neu entstehende Hülle einlagern oder ein Virus kann gänzlich mit der Hülle des anderen Virus verpackt werden. Dieses Phänomen der Pseudotypen-Entstehung wird auch als Phänotypische Mischung („Phenotypic mixing“) bezeichnet.[49]
Virushülle und Virusinaktivierung
Der Verlust der Virushülle oder die Entfernung der Lipidkomponenten aus der Hülle verhindern, dass das behüllte Virus die Wirtszelle infizieren kann. Dieser Umstand wird zur Inaktivierung von behüllten Viren genutzt, um eine Verbreitung des Virus zu unterbinden. Die empfindlichste Komponente der Virushülle, die Lipidmembran, kann durch fettlösende Alkohole wie Ethanol oder 2-Propanol zerstört werden.[50] Bei einem hohen Lipidanteil der Virushülle wie bei den Orthomyxoviren genügen schon milde Detergenzien oder Seifen, um die Infektiosität des Virus herabzusetzen. Bei der Inaktivierung von möglichen behüllten Viren wie HIV, HBV und HCV in Blutprodukten zur Transfusion kann eine Kombination von milden Lösungsmitteln und Detergenzien verwendet werden.[51]
Entstehung von Pandemien und „neuen Viren“
Die hohe immunologische Flexibilität der Hüllproteine erlaubt es einigen behüllten Viren, sich in verschiedenen Wirtsspezies vermehren zu können. So können Infektionen artübergreifend neu entstehen oder Zwischenwirte als Überträger genutzt werden. Die von Gliederfüßern (beispielsweise Stechmücken und Zecken) übertragenen Viren, die sogenannten Arboviren, sind daher überwiegend behüllte Viren. Die einzige unbehüllte Gattung Coltivirus, deren Mitglieder als Arboviren übertragen werden können, besitzen als Ersatz für die Flexibilität der Virushülle ein zweites Kapsid. Viren sind meist dann besonders pathogen, wenn sie in einer Wirtspopulation neu auftreten. Daher haben die behüllten Viren, die den Wirtsübergang vom Tier zum Mensch besonders begünstigen, ein besonders hohes Potential für neu beim Menschen auftretende Infektionen.
Literatur
- Stephen C. Harrison: Principles of Virus Structure. In: David M. Knipe, Peter M. Howley et al. (eds.): Fields’ Virology. 4. Auflage. Philadelphia 2001, ISBN 0-7817-1832-5, S. 53–85
- John A. T. Young: Virus Entry and Uncoating. In: Fields’ Virology.
- S. J. Flint, L. W. Enquist, V. R. Racaniello und A. M. Skalka: Principles of Virology. Molecular Biology, Pathogenesis, and Control of Animal Viruses. 2. Auflage. ASM-Press, Washington DC 2004, ISBN 1-55581-259-7
- Joe Bentz (ed.): Viral Fusion Mechanisms. CRC-Press, Boca Raton 1993, ISBN 0-8493-5606-7
- Robert Brasseur (ed.): Molecular Description of Biological Membranes by Computer Aided Conformational Analysis. Vol. 1, CRC-Press, Boston 1990, ISBN 0-8493-6375-6
- Wolfhard Weidel: Virus – Die Geschichte vom geborgten Leben. Berlin / Göttingen / Heidelberg 1957
- Frank Fenner, B. R. McAuslan et al. (eds.): The Biology of Animal Viruses. Academic Press, New York / London, 1. Auflage 1968, 2. Auflage 1974, ISBN 0-12-253040-3
- Alena Lengerová: Membrane Antigens. Fischer, Jena 1977
Einzelnachweise
- ↑ Karlheinz Lüdtke:
 Zur Geschichte der frühen Virusforschung. (PDF; 217 kB),
MPI für Wissenschaftsgeschichte, 1999 (Übersicht)
Zur Geschichte der frühen Virusforschung. (PDF; 217 kB),
MPI für Wissenschaftsgeschichte, 1999 (Übersicht)
- ↑ W. Reed: Recent researches concerning the etiology, propagation and prevention of yellow fever by the United States Army Commission. In: J. Trop. Med., 1901, 5, S. 143–158
- ↑ B. v. Borries, E. Ruska, H. Ruska: Bakterien und Virus in übermikroskopischer Aufnahme. Klin. Wochenschrift, 1938, 17, S. 921–925
- ↑ H. Ruska: Versuch zu einer Ordnung der Virusarten. In: Arch. ges. Virusforsch., 1943, 2, S. 480–498
- ↑ E. W. Goodpasture, A. M. Woodruff, G. J. Buddingh: Vaccinal infection of the chorio-allantoic membrane of the chick embryo. In: Amer. J. Pathol., 1932, 8, S. 271
- ↑ W. I. B. Beverige, F. M. Burnet: The cultivation of viruses and rickettsiae in the chick embryo. In: Med. Res. Council Spec. Rept. Ser, 1946, S. 256
- ↑ W. B. Dunham, W. J. Macneal: Culture on the Chick Chorio-allantois as a Test of Inactivation of Vaccinia Virus.
In: J. Bacteriology, 1942, 44(4), S. 413–424, PMID 16560579
- ↑ M. Uhler, S. Gard: Lipid content of standard and incomplete influenza A virus. In:
Nature 1954, 173(4413), S. 1041–1042, PMID 13165714
- ↑ E. Gorter, F. Grendel: On bimolecular layers of lipoid on the chromocytes of the blood. In: J. Exp. Med.,
1925, 41, S. 439–443, jem.org
- ↑ L. H. Frommhagen, N. K. Freeman, C. A. Knight: The lipid constituents of influenza virus, chick allantoic
membrane and sedimentable allantoic protein. In: Virology, 1958, 5(1), S. 173–175,
PMID 13519759
- ↑ L. L. Coriell, G. Rake et al.: Electron microscopy of herpes simplex. In: J Bacteriol., 1950, 59(1),
S. 61–68, PMID 15400321
- ↑ S. Brenner, R. W. Horne: A negative staining method for high resolution electron microscopy of viruses. In:
Biochim. Biophys. Acta, 1959, 34, S. 103–110, PMID 13804200
- ↑ D. Branton: Fracture faces of frozen membranes. In: PNAS, 1966, 55, S. 1048–1056, PMID 5334198
- ↑ S. J. Singer, G. L. Nicholson: The fluid mosaic model of the structure of cell membranes. In:
Science, 1972, 175, S. 720–731, PMID 4333397
- ↑ J. W. Corran, W. C. Lewis: Lecithin and Cholesterol in Relation to the Physical Nature of Cell Membranes.
In: Biochem J., 1924, 18(6), S. 1364–1370, PMID 16743417
- ↑ R. P. Rand, V. Luzzati: X-ray diffraction study in water of lipids extracted from human erythrocytes: the
position of cholesterol in the lipid lamellae. In: Biophys. J., 1968, 8(1), S. 125–137,
PMID 5641398
- ↑ J. P. Laliberte, L. W. McGinnes et al.: Integrity of membrane lipid rafts is necessary for the ordered assembly
and release of infectious Newcastle disease virus particles. In: J. Virol. 2006, 80(21), S. 10652–10662,
PMID 17041223
- ↑ H. Imhoff, V. von Messling et al.: Canine distemper virus infection requires cholesterol in the viral
envelope. In: J. Virology, 2007, 81(8), S. 4158–4165, PMID 17267508
- ↑ S. Hambleton et al.: Cholesterol dependence of varicella-zoster virion entry into target cells. In:
J. Virology, 2007, 81(14), S. 7548–7558, PMID 17494071
- ↑ M. K. Jain, R. C. Wagner: Introduction to Biological Membranes. Wiley, New York 1980, ISBN 0-471-03471-1.
- ↑ in molare Prozent umgerechnet aus: R. C. Aloia et al.: Lipid composition and fluidity of the human
immunodeficiency virus. In: PNAS, 1988, 85(3), S. 900–904, PMID 2829209
- ↑ berechnet aus Tabelle 1 und 4 in: O. Satoh et al.: Membrane structure of the hepatitis B virus surface antigen
particle. In: J. Biochemistry Tokyo, 2000, 127(4), S. 543–550, PMID 10739944
- ↑ G. van Meer, K. Simons: Viruses budding from either the apical or the basolateral plasma membrane domain of
MDCK cells have unique phospholipid compositions. In: EMBO J., 1982, 1(7), S. 847–852, PMID 6329709
- ↑ N. Chazal, D. Gerlier: Virus Entry, Assembly, Budding, and Membrane Rafts. In: Microbiol. Mol. Biol.
Rev., 2003, 67(2), S. 226–237 (Review PMID 12794191)
- ↑ B. A., G. Meyers: The pestivirus glycoprotein Erns is anchored in plane in the membrane via an
amphipathic helix. In: J. Biol. Chem., 2007, E-pub,
PMID 17848558
- ↑ B. Rentier, E. L. Hooghe-Peters, M. Dubois-Dalcq: Electron microscopic study of measles virus infection:
cell fusion and hemadsorption. J. Virol., 1978, 28(2), S. 567–577,
PMID 722861
- ↑ J. Torres, K. Parthasarathy et al.: Model of a putative pore: the pentameric alpha-helical bundle of
SARS coronavirus E protein in lipid bilayers. In: Biophys. J., 2006, 91(3), S. 938–947,
PMID 16698774
- ↑ R. H. Cheng, R. J. Kuhn et al.: Nucleocapsid and glycoprotein organization in an enveloped virus. In:
Cell, 1995, 80(4), S. 621–630, PMID 7867069
- ↑ R. H. Vogel, S. W. Provencher et al.: Envelope structure of Semliki Forest virus reconstructed from cryo-electron
micrographs. In: Nature, 1986, 320(6062), S. 533–535, PMID 3960136
- ↑ C. H. von Bonsdorff, R. Pettersson: Surface structure of Uukuniemi virus. In: J. Virology, 1975, 16(5),
S. 1296–1307, PMID 52726
- ↑ C. Risco et al.: Endoplasmic reticulum-Golgi intermediate compartment membranes and vimentin filaments
participate in vaccinia virus assembly. In: J. Virol., 2002, 76(4), S. 1839–1855, PMID 11799179
- ↑ als Übersicht zur Virusreifung und Knospung siehe: H. Garoff, R. Hewson,D.-J. E. Opstelten: Virus maturation by
budding. In: Microbiol. Mol. Biol. Rev., 1998, 62, S. 1171–1190, PMID 9841669
- ↑ S. Tzlil, M. Deserno et al.: A statistical-thermodynamic model of viral budding. In: Biophys. J., 2004,
86(4), S. 2037–2048, PMID 15041646
- ↑ D. M. Lerner, J. M. Deutsch, G. F. Oster: How does a virus bud? Biophys J., 1993, 65(1), S. 73–79,
PMID 8369463
- ↑ H. Garoff, K. Simons: Location of the spike glycoproteins in the Semliki Forest virus membrane. In:
PNAS, 1974, 71(10), S. 3988–3992, PMID 4530279
- ↑ M. Delchambre et al.: The GAG precursor of simian immunodeficiency virus assembles into virus-like particles.
In: EMBO J., 1989, 8(9), S. 2653–2660, PMID 2684654
- ↑ T. L. Cadd, U. Skoging, P. Liljeström: Budding of enveloped viruses from the plasma membrane. In:
Bioessays, 1997, 19(11), S. 993–1000, PMID 9394621
- ↑ D. Falke, R. Siegert, W. Vogell: Elektronenmikroskopische Befunde zur Frage der Doppelmembranbildung des
Herpes-simplex-Virus. In: Arch Gesamte Virusforschung 1959, 9, S. 484–496, PMID 13821428
- ↑ I. L. van Genderen, R. Brandimarti et al.: The phospholipid composition of extracellular herpes simplex virions
differs from that of host cell nuclei. In: Virology, 1994, 200(2), S. 831–836, PMID 8178468
- ↑ L. M. Stannard et al.: Electron microscopic study of the distribution of the Australia antigen in individual
sera of 50 serologically positive blood donors and two patients with serum hepatitis. In: J. Clin. Pathol., 1973, 26(3), S. 209–216,
PMID 4700502
- ↑ CG Schüttler et al.: Variable ratio of hepatitis C virus RNA to viral core antigen in patient sera.
In: J. Clin. Microbiol., 2004, 42(5), S. 1977–1981, PMID 15131157
- ↑ F. Bonino, K. H. Heermann, M. Rizzetto, W. H. Gerlich: Hepatitis delta virus: protein composition of delta
antigen and its hepatitis B virus-derived envelope. In: J. Virology, 1986, 58(3), S. 945–950, PMID 3701932
- ↑ R. A. Crowther, N. A. Kiselev et al.: Three-dimensional structure of hepatitis B virus core particles determined
by electron cryomicroscopy. In: Cell, 1994, 77(6), S. 943–950, PMID 8004680
- ↑ E. L. Oleszak et al.: Molecular mimicry between Fc receptor and S peplomer protein of mouse hepatitis virus,
bovine corona virus, and transmissible gastroenteritis virus. In: Hybridoma, 1995, 14(1), S. 1–8,
PMID 7768529
- ↑ Y. W. Hu et al.: Immunoglobulin mimicry by Hepatitis C Virus envelope protein E2. In: Virology,
2005, 332(2), S. 538–549, PMID 15680419
- ↑ J. A. Quiroga et al.: Inhibition of albumin binding to hepatitis B virions by monoclonal antibody to the
preS2 domain of the viral envelope. In: Digestion, 1987, 38(4), S. 212–220, PMID 2452108
- ↑ P. F. Serres: Molecular mimicry between the trimeric ectodomain of the transmembrane protein of immunosuppressive
lentiviruses (HIV-SIV-FIV) and interleukin 2. In: C R Acad. Sci. III, 2000, 323(11), S. 1019–1029, PMID 11144025
- ↑ J. A. Briggs, T. Wilk, S. D. Fuller: Do lipid rafts mediate virus assembly and pseudotyping?
J. General Virology, 2003, 84(Pt 4), S. 757–768 (Review PMID 12655075)
- ↑ J. Dragunova et al.: Phenotypic mixing between vesicular stomatitis and Uukuniemi viruses. In:
Acta Virol., 1986, 30(6), S. 512–514, PMID 2881472
- ↑ W. R. Moorer: Antiviral activity of alcohol for surface disinfection. Int. J. Dent. Hyg., 2003) 1(3),
S. 138–142 (Review PMID 16451513)
- ↑ B. Horowitz et al.: Viral safety of solvent-detergent treated blood products. In: Dev. Biol. Stand.,
1993, 81, S. 147–161 (Review PMID 8174797)


© biancahoegel.de
Datum der letzten Änderung: Jena, den: 29.05. 2026