Aromatizität
Aromatizität ist ein Konzept aus dem Bereich der chemischen Bindung, das eingeführt wurde, um die auffallenden Eigenschaften der Stoffklasse der Aromaten zu erklären. Es beschreibt das Phänomen, dass cyclische Delokalisation von 4n + 2 π-Elektronen (Hückel-Regel) in einem planaren Molekül zu einer energetischen Stabilisierung führt, die mit dem Auftreten von diamagnetischen Ringstromeffekten, Anisotropie der magnetischen Suszeptibilität, magnetic susceptibility exaltation sowie einer Tendenz zum Bindungslängenausgleich einhergeht. Das Phänomen der Aromatizität ist keine summarische Observable, hingegen sind die einzelnen Kriterien messbar. Jedoch sind für die einzelnen Kriterien keine festen oder klaren Grenzwerte zur Abgrenzung aromatisch – nichtaromatisch - antiaromatisch festgelegt. Üblicherweise erfolgt die Diskussion über das Ausmaß der Aromatizität einzelner Verbindungen unter Bezug auf eine Referenz. Hierbei gilt Benzol unumstritten als Paradebeispiel für ein aromatisches System.
Während sich ursprünglich die Diskussion der Aromatizität auf cyclisch konjugierte π-Systeme beschränkt hat, ist der Aromatizitätsbegriff heute deutlich weiter gefasst und um Homoaromatizität, σ-Aromatizität, σ-Homoaromatizität, räumliche Aromatizität, Superaromatizität etc. ergänzt. Insbesondere die Entwicklung quantenchemischer Methoden hat dabei die Erweiterung des theoretischen Verständnisses signifikant unterstützt.
Sind in einem Molekül statt 4n + 2 Elektronen 4n Elektronen cyclisch delokalisiert, so beobachtet man das Phänomen der Antiaromatizität. In diesem Fall erfolgt eine Destabilisierung des Systems. Antiaromatische Systeme werden somit versuchen, der ungünstigen Situation zu entgehen und durch geometrische Veränderung (Verzerrung, Jahn-Teller-Effekt) die cyclische Delokalisation zu verringern. Ein Paradebeispiel hierfür ist Cyclobutadien.
Eine Übersicht der Charakteristika von aromatischen und antiaromatischen Systemen im Vergleich zu linear-konjugierten Referenzen fasst die nachfolgende Tabelle zusammen:
| Eigenschaft | Aromat | Referenz (Olefin) | Antiaromat |
|---|---|---|---|
| Delokalisation / Konjugation | cyclisch | linear | cyclisch |
| Zahl der π-Elektronen | 4n + 2 | 2n | 4n |
| Energetischer Effekt der Konjugation | Stabilisierung | = Referenz | Destabilisierung |
| Ausmaß der Delokalisation | erhöht | = Referenz | erniedrigt |
| Bindungslängen | Tendenz
zum Bindungslängenausgleich |
alternierend | alternierend |
| Diamagnetische Anisotropie | erhöht | - | klein |
| magnetic susceptibility exaltation | hoch | - | niedrig |
| Ringstrom | diamagnetisch | - | paramagnetisch |
| NICS-Werte | deutlich negativ | - | deutlich positiv |
| Chemische Reaktivität | elektrophile Substitution | Addition | Addition |
| HOMO-LUMO-Differenz | erhöht | = Referenz | erniedrigt |
| Typische Vertreter | Benzol | Butadien | Cyclobutadien |
Entwicklung des Aromatizitätsbegriffs
Aromatizität wurde zunächst im Sinne olfaktorischer und chemischer Eigenschaften beschrieben: besonders „aromatisch“ riechende Verbindungen zeigten für ungesättigte Verbindungen verblüffend geringe Reaktivität. So wird z.B. Brom nicht spontan an die Doppelbindungen von Benzol addiert. Benzol und seine Derivate reagieren zudem bevorzugt nach dem Muster der elektrophilen aromatischen Substitution (Additions-Eliminierungsmechanismus, der zur Aufrechterhaltung der cyclischen Delokalisation führt) und nicht unter einfacher Addition an eine der (formalen) Doppelbindungen. Während sich frühe Arbeiten auf synthetische Methoden stützten – z.B. Versuche, über die Zahl an Isomeren die cyclische Struktur von Benzol nachzuweisen und die Frage zu klären, ob Alternanz von Doppel- und Einfachbindungen vorliegt – wurden im Zuge der Entwicklung der (Elektronen-)Theorie der chemischen Bindung mehr und mehr physikalische Aspekte in die Untersuchungen einbezogen. Eine erschöpfende Zusammenstellung wichtiger Meilensteine der Entwicklung des Aromatizitätsbegriffs und der Kriterien zur Definition von Aromatizität findet sich bei Schleyer et al. – einige Kernaspekte können der nachfolgenden Tabelle entnommen werden:
| Jahr | Aspekt |
|---|---|
| 1865 | Kekulé: Benzolstruktur |
| 1866 | Erlenmeyer: Vorherrschen von elektrophiler Substitution statt Addition |
| 1910 | Pascal: Aromaten haben erhöhte diamagnetische Suszeptibilität |
| 1925 | Armit & Robinson: Elektronensextett und Heteroaromatizität |
| 1931 | Hückel: Stabilisierung der 4n + 2-, Destabilisierung der 4n-Elektronensysteme (Hückel-Regel) |
| 1936 / 1937 | London, Pauling: Theorie der Ringstromeffekte |
| 1956 | Pople: Einfluss von Ringstromeffekten auf NMR-Verschiebungen |
| 1969 / 1970 | Dauben / Benson & Flygare: magnetische Suszeptibilität (Erhöhung und Anisotropie) als Aromatizitätskriterien |
| 1979 | Aihara: Dreidimensionale Aromatizität |
| 1980 | Kutzelnigg: Quantenchemische Berechnung (IGLO) von NMR- und magnetischen Moleküleigenschaften |
| 1984 | Dewar: σ-Aromatizität von Cyclopropan |
| 1996 | Schleyer: Einführung von NICS als einfache quantenchemische Methode zur Untersuchung der Aromatizität |
Für eine Übersicht der frühen Geschichte der Untersuchung der Aromaten vgl. z. B. unter Benzol.
Arten der Aromatizität
- π-Aromatizität
Beim archetypischen Fall von Aromatizität findet cyclische Delokalisation durch Konjugation von π-Orbitalen entlang eines geschlossenen Perimeters statt, der von σ-Bindungen definiert wird.
- Hetero-Aromatizität
Ausgehend von benzoiden Aromaten werden einzelne Kohlenstoff-Atome durch Heteroatome ersetzt. Pyridin, Thiophen, Furan, Pyrrol sind typische Beispiele.
- σ-Aromatizität
Bei kleinen Ringen (insbesondere bei Cyclopropanen) wird cyclische Delokalisation in der σ-Ebene beobachtet. Wie bei der π-Aromatizität werden energetische Stabilisierung und die für diamagnetische Ringströme typischen magnetischen Eigenschaften beobachtet. Bei rein anorganischen Systemen gilt das cyclische Ion H3+ als Paradebeispiel für σ-Aromatizität. Auch für trigonale Metallcluster von Zn und Au wird σ-Aromatizität diskutiert.
- Homoaromatizität
Wird im Falle der π-Aromaten der geschlossene Perimeter aus σ-Bindungen an einer Stelle unterbrochen, z. B. durch Einführung einer Methylen-Gruppe, so kann an dieser Position bei ausreichenden geometrischen Bedingungen die π-Konjugation durch den Raum (through space) aufrechterhalten werden. Paradebeispiel für dieses Phänomen ist das Homotropylium-Kation. Ist der Perimeter an mehreren Stellen unterbrochen, spricht man von Bishomo-, Trishomo-Aromatizität usw.
- σ-Homoaromatizität
Wie bei den π-Aromaten kann bei kleinen Ringen die σ-Konjugation durch den Raum erfolgen (through space). Beispiele für σ-Bishomoaromatizität in 2- und 6-Elektronensystemen sind beschrieben.
- Sphärische Aromatizität (Räumliche Aromatizität, 3D-Aromatizität)
Aromatizität in drei Dimensionen ist beschrieben für Cluster-Verbindungen wie Borane und Fullerene.
Aromatische Stabilisierungsenergie
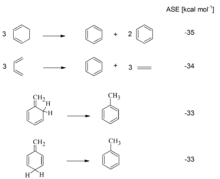
Eine früh bekannte Auffälligkeit von Benzol war die Tatsache, dass sich Benzol – im Gegensatz zu anderen ungesättigten Verbindungen – nur vergleichsweise schwer hydrieren lässt. Bei thermochemischen Messungen fiel zudem auf, dass die im Falle von Benzol freiwerdende Hydrierwärme gegenüber einem hypothetischen nichtaromatischen Cyclohexatrien signifikant reduziert ist. Aus dieser Differenz wurde eine aromatische Stabilisierungsenergie des Benzols (ASE) von −150 kJ/mol (−36 kcal/mol) abgeleitet. (Die Vorzeichenkonvention der ASE wird in der Literatur leider nicht immer stringent eingehalten. In diesem Artikel wird für aromatische Verbindungen eine negative ASE (Stabilisierung!) und eine positive ASE (Destabilisierung!) für antiaromatische Verbindungen verwendet.)
Neben thermochemischen Messungen stehen heute eine Vielzahl von quantenchemischen Methoden zur Verfügung, um aromatische Stabilisierungsenergien abzuschätzen. Üblicherweise geschieht dies anhand von homodesmotischen Reaktionen. Durch Verwendung von homodesmotischen Reaktionsgleichungen sollen sich dabei Ungenauigkeiten, die sich aus der Qualität der verwendeten Rechenmethoden ergeben, herausmitteln, so dass die interessierende Größe in hoher Genauigkeit erhalten wird. Unabhängig davon, ob die Abschätzung der aromatischen Stabilisierungsenergie nun über thermochemische Messungen oder Berechnungen erfolgt, muss stets beachtet werden, dass die aromatische Stabilisierungsenergie keine Observable ist. Die ASE hängt damit von den gewählten Referenzverbindungen ab und ist zudem nicht leicht von anderen Effekten – etwa Ringspannungen – zu trennen. Auch müssen Stabilisierungseffekte durch Resonanz herausgerechnet werden, die nicht auf der cyclischen Delokalisation beruhen und somit zwar zur gesamten Resonanzenergie beitragen, aber nicht zur ASE. Diese Probleme zeigen sich bereits deutlich bei der Berechnung der ASE von Benzol: selbst bei sorgfältiger Auswahl der Referenzstrukturen für homodesmotische Reaktionen schwanken die berechneten Werte leicht um 10 kcal mol−1, wie eine Zusammenstellung von Cyrański zeigt. Um das Problem zu umgehen, schlugen Schleyer et al. vor, Isomerisierungsreaktionen anstelle von homodesmotischen Reaktionsschemata zur Ermittlung der ASE heranzuziehen. Die berechneten isomerization stabilization energies (ISE) sind dabei in guter Übereinstimmung mit Vergleichswerten aus sorgfältig ausgewählten homodesmotischen Reaktionsschemata.
Eine Reihe von Schleyer et al. anhand einer "homodesmotischen" Reaktion berechneten ASEs für 5-gliedrige Ringverbindungen belegen sowohl die Hückel-Regel als auch das schon früh bekannte Phänomen der Heteroaromatizität:
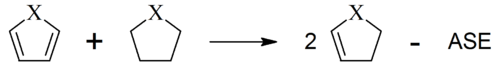
| ASE [kcal mol−1] | |||||||
|---|---|---|---|---|---|---|---|
| X | CH | CH2 | SiH | BH | NH | O | S |
| Name von C4H4X | Cyclopentadien | Borol | Pyrrol | Furan | Thiophen | ||
| Kation | 57 | 24 | |||||
| Neutralverbindung | −4 | 19 | −26 | −20 | −22 | ||
| Anion | −29 | −14 | |||||
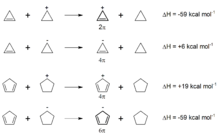
Das Kriterium der aromatischen Stabilisierungsenergie folgt der "praktischen Erfahrung" der Chemiker, dass sich aromatische Verbindungen durch besondere Stabilität auszeichnen (negativer Wert für die ASE). Die oben angeführten Werte für antiaromatische Systeme (Tabelle, 5-Ring-Systeme mit 4n π-Elektronen) sind hiermit in Einklang (Destabilisierung durch Antiaromatizität). Cyclobutadien, ein Paradefall für Antiaromatizität, erfährt nach der Abschätzung
- Cyclobutadien + Cyclobutan → 2 Cyclobuten
eine Destabilisierung von +36 kcal/mol.
Eindrückliche Beispiele dafür, dass cyclische Konjugation von 4n + 2 Elektronen zu einer Stabilisierung, cyclische Konjugation von 4n Elektronen dagegen zu einer Destabilisierung führt, liefern isodesmische Reaktionen von Cyclopropen bzw. Cyclopentadien mit Cyclopropyl- bzw. Cyclopentyl-Anionen und -Kationen (vgl. nebenstehende Abbildung). Die Bildung von aromatischen Ionen wird dabei durch hohe Exothermie, die Bildung von antiaromatischen Ionen durch deutliche Endothermie angezeigt.
NMR-Effekte, NICS
In NMR-Experimenten lässt sich der Effekt der cyclischen Elektronendelokalisation in aromatischen Verbindungen durch die damit einhergehenden magnetischen Effekte (Ringstromeffekt) nachweisen: das durch den Ringstrom induzierte magnetische Feld führt bei Benzol zu einer Protonenresonanz bei 7,3 ppm (Entschirmung), wohingegen vergleichbare vinylische Protonen in Cyclohexen bei 5,6 ppm erscheinen. Protonen, welche sich oberhalb oder im inneren Bereich eines aromatischen π-Systems befinden, erfahren dagegen eine Abschirmung. Lehrbuchbeispiele für diesen Effekt sind z.B. die Methylenprotonen im Methano[10]annulen (Vogel-Aromat) oder die inneren Protonen im [18]Annulen.
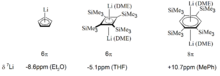
Neben den Protonenresonanzen wurden auch die chemischen Verschiebungen von Li+ häufig zum Studium der aromatischen Eigenschaften anionischer π-Systeme herangezogen, da Lithium in der Regel in zentraler Position oberhalb des π-Systems elektrostatisch bindet und somit eine wenig störanfällige Probe für die magnetischen Eigenschaften der zu untersuchenden Spezies darstellt. Typische Beispiele für diese Herangehensweise finden sich in den Li-Salzen der (Di)Anionen von Cyclopentadien, Tetrakis-trimethylsilyl-cyclobutadien und dem antiaromatischen Dianion von Tetrakis-trimethylsilyl-benzol (vgl. Abbildung).
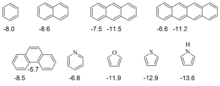
Mit der Einführung quantenchemischer Methoden wurde es möglich, chemische Verschiebungen (Abschirmungen) an einer beliebigen Stelle im Raum zu berechnen und somit Ringstromeffekte unabhängig von einzelnen Atomkernen zu evaluieren. Die Methode wurde von ihren Protagonisten Nucleus Independent Chemical Shifts (NICS) benannt. Zum Studium aromatischer Verbindungen werden NICS-Werte in der Regel im geometrischen Mittelpunkt der Schweratome (d. h. der nicht-Wasserstoffatome, nicht massengemittelt) berechnet. Wie im Falle von Protonen gewohnt, zeigen negative NICS-Werte im inneren von cyclisch delokalisierten Systemen eine diamagnetischen Ringstrom und somit Aromatizität an. Positive NICS-Werte indizieren dagegen Antiaromatizität.
Obwohl die cyclische Elektronendelokalisation die magnetischen Eigenschaften (anti)aromatischer Moleküle dominiert, zeigen auch lokalisierte Bindungen und lone pairs zumindest kurzreichweitige magnetische Effekte, welche die berechneten NICS-Werte beeinflussen können. Um diesen Störfaktor auszuschalten, können bei planaren Molekülen NICS-Werte oberhalb der π-Ebene berechnet werden. Aufwändigere Methoden erlauben es ferner, NICS-Beiträge von σ- und π-Bindungen oder auch von einzelnen Molekülorbitalen zu berechnen und somit zu separieren.
Magnetische Suszeptibilität
Die magnetische Suszeptibilität χ ist eine aus der Physik bekannte Größe und beschreibt, wie stark sich ein Stoff beim Einbringen in ein Magnetfeld magnetisieren lässt. Die magnetische Suszeptibilität lässt sich auf Beiträge entlang der drei Raumrichtungen (x, y, z) zurückführen und wird in der Chemie meist als molare Größe verwendet. Für ungeordnete Flüssigkeiten ergibt sich dabei die beobachtbare Suszeptibilität als Mittelwert aus den drei Suszeptibilitäten der Moleküle entlang der Raumrichtungen:
- χM = 1/3 (χx + χz + χz)
Orientiert man benzoide Aromaten derart, dass das Molekül in der xy-Ebene zu liegen kommt, so ist die magnetische Suszeptibilität in z-Richtung deutlich größer als innerhalb der Molekülebene. Dieses Phänomen wird als erhöhte diamagnetische Anisotropie bezeichnet und durch folgende Gleichung beschrieben:
- Δχ = χz − 1/2 (χx + χy) >> 0
Da die experimentelle Bestimmung der anisotropen Eigenschaften schwierig ist und nur an Einkristallen vorgenommen werden kann, wurden einfachere Kriterien zur Beschreibung der magnetischen Eigenschaften von organischen Molekülen zu Studium der Aromatizität gesucht. Einen eleganten Ansatz schlugen Dauben, Wilson und Laity vor: ähnlich wie sich thermodynamische Größen für Moleküle aus entsprechenden Inkrementen für die enthaltenen Bindungen abschätzen lassen, können magnetische Suszeptibilitäten auf Bindungs-spezifische Beiträge zurückgeführt werden. Im Falle von aromatischen Verbindungen stellt man dabei fest, dass die gemessenen Suszeptibilitäten gegenüber den aus Inkrementen abgeschätzten Werten erhöht sind. Dieser Effekt wird auf die besonderen magnetischen Effekte durch die cyclische Delokalisation zurückgeführt. Für den Effekt wurde die Bezeichnung magnetic susceptibility exaltation eingeführt gemäß der Gleichung
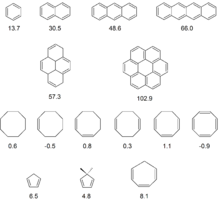
- Λ = χM (gemessen) − χM (Inkrement)
Für aromatische Moleküle gilt Λ >> 0, nicht-aromatische Moleküle zeigen Λ ~ 0. Da antiaromatische Moleküle versuchen, die cyclische Delokalisation durch geometrische Veränderung zu minimieren, werden keine signifikant negativen Λ-Werte ermittelt (vgl. Cyclooctatetraen).
Anhand der in der nebenstehenden Abbildung aufgeführten Werte für "klassische" Aromaten lässt sich ableiten, dass diese sich durch Λ > 12 auszeichnen und die Größe von Λ mit der Größe der π-Systeme korreliert. Die Reihe Cyclooctan → Cyclooctatetraen belegt, dass die einfache Einführung von Doppelbindungen sowie deren nicht-cyclische Konjugation nur vernachlässigbaren Einfluss auf die Größe von Λ haben. Cyclooctatetraen schließlich weicht dem energetisch ungünstigen antiaromatischen Zustand durch geometrische Verzerrung weitgehend aus, so dass zwar ein negativer Wert für Λ ermittelt wird, dieser fällt jedoch mit −0,9 überraschend gering aus. Interessante Sonderfälle werden bei Cyclopentadien und 5,5-Dimethylcyclopentadien sowie bei Cycloheptatrien angetroffen. Die auffallend hohen Werte der magnetischen Suszeptibilitätserhöhung wurden bei den Cyclopentadienen als Hinweis auf aromatischen Charakter durch Einbeziehung von zwei Elektronen aus den CH/CC-Bindungen der Methyleneinheit interpretiert, der hohe Λ-Wert von Cycloheptatrien ist in Einklang mit dem Effekt der Homoaromatizität.
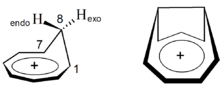
Eine zeitweilige Renaissance hat die Betrachtung magnetischer Suszeptibilitäten durch Einführung leistungsfähiger quantenmechanischer Methoden erfahren. So nutzten Cremer et al. berechnete magnetische Suszeptibilitäten zum Nachweis der homoaromatischen Natur des Homotropylium-Kations. Bei Variation der C1-C7-Bindungslänge zeigt das Kation in seiner Gleichgewichtsgeometrie die höchste magnetische Suszeptibilität. Der Effekt geht einher mit der größten Differenz der chemischen Verschiebungen von H8-endo und H8-exo, der größten Angleichung der C1–C7 13C-Resonanzen sowie dem stärksten Bindungslängenausgleich zwischen den an der cyclischen Delokalisation beteiligten Kohlenstoff-Atomen. Diese Untersuchungen wurden unter anderem auf ein bishomoaromatisches 1,4-Bishomotropylium-Kation ausgedehnt.
Herges und Schleyer nutzten quantenchemisch berechnete magnetische Suszeptibilitäten und deren Anisotropien, um den aromatischen Charakter von Übergangszuständen von pericyclischen Reaktionen wie z. B. der Diels-Alder-Reaktion nachzuweisen. Die Berechnung von magnetischen Suszeptibilitäten und ihren Anisotropien ist heute jedoch von anderen Methoden wie der Berechnung von NICS-Werten oder auch der Visualisierung und Quantifizierung von induzierten Ringströmen abgelöst worden, die einen detaillierteren Einblick in das Phänomen der Aromatizität geben.
Geometrische Effekte
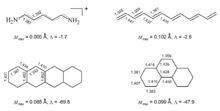
Als charakteristisches Merkmal der cyclischen Elektronendelokalisation wird immer wieder – ausgehend vom "klassischen" Fall der π-Aromatizität – der Bindungslängenausgleich zwischen Einfach- und Doppelbindungen herausgestellt. Benzol, als Archetypus der Aromaten, zeigt dieses Phänomen in Perfektion, ebenso wie Borazin, das allenfalls als Grenzfall für Aromatizität gelten kann (starke Lokalisation der Elektronen an den Stickstoffatomen, geringer Ringstrom). Schleyer et al. weisen darauf hin, dass die Bindungslängendifferenzen in polycyclischen Aromaten sich nicht signifikant von denen linear konjugierter Polyene unterscheiden müssen und unter Umständen sogar in den linearen Systemen kleiner ausfallen können (vgl. Abbildung). Bindungslängenausgleich ist somit für sich allein genommen kein hinreichendes Kriterium für aromatischen Charakter.


© biancahoegel.de
Datum der letzten Änderung: Jena, den: 02.09. 2025