Destillation
Destillation (lat. destillare „herabtröpfeln“) ist ein thermisches Trennverfahren, um verdampfbare Flüssigkeiten zu gewinnen oder Lösungsmittel von schwer verdampfbaren Stoffen abzutrennen. Die Destillation hat gegenüber anderen Trennverfahren den Vorteil, dass in der Regel keine weiteren Stoffe wie Adsorbentien oder Lösungsmittel hinzugefügt werden.
Bei der Destillation wird zunächst das Ausgangsgemisch zum Sieden gebracht. Der entstehende Dampf, der sich aus den verschiedenen flüchtigen Komponenten der zu trennenden Lösung zusammensetzt, wird in einem Kondensator durch Abkühlen wieder verflüssigt. Im Labormaßstab wird oft der Liebigkühler verwendet. Im Anschluss wird das flüssige Kondensat aufgefangen. Typische Anwendungen der Destillation sind das Brennen von Alkohol und das Destillieren (die Rektifikation) von Erdöl in der Raffinerie oder auch die Herstellung von destilliertem Wasser.
Idealerweise soll sich die Flüssigkeit beim Destillieren nicht zersetzen. Bei der sogenannten „trockenen Destillation“ ist dies anders: dabei werden nicht-verdampfbare feste Stoffe in kleinere Moleküle zerlegt. So erhielt man z. B. bei der trockenen Destillation von Holz den „Holzgeist“ (Methanol). Zwar wird hier eine verdampfbare Flüssigkeit durch Kondensation gewonnen, aber nach der heutigen Theorie liegt hier keine destillative Trennung vor. Daher nennt man dieses Verfahren besser Thermolyse oder Pyrolyse.
Geschichte


Die Destillation von Pech und Teer zur Abdichtung von Schiffen, als Klebemittel und auch als Heilmittel ist seit der Jungsteinzeit bekannt; sie wurde mit einfachsten Mitteln bewerkstelligt. In der Antike wurden vor allem ätherische Öle, als Riech- und Duftstoffe, destilliert. Die ältesten bei archäologischen Ausgrabungen aufgefundenen Darstellungen von Destilliergeräten stammen aus Mesopotamien, dem heutigen Irak, und werden auf ein Alter von über 5500 Jahren geschätzt. Diese ersten Geräte bestanden aus einem Gefäß mit einem Deckel, an dem sich beim Erhitzen das Destillat niederschlug. Damit diese Flüssigkeit nicht wieder in das Gefäß zurück tropfte, verwendete man im Deckel Schwämme oder Wollbüschel, um die Flüssigkeit aufzusaugen. Diese wurden dann einfach regelmäßig ausgepresst, um das Destillat zu erhalten.
Mit derselben Methode erzeugten griechische Seefahrer um 500 v. Chr. Trinkwasser aus Meerwasser. Aristoteles beschrieb unter anderem im 4. Jahrhundert vor Christus, wie Meerwasser durch Destillation trinkbar gemacht werden kann. Er beschrieb weiter, dass Weine und andere Flüssigkeiten demselben Verfahren unterzogen werden können. Etwa 200 vor Christus erklärte Alexander von Aphrodisias das Verfahren, destilliertes Wasser herzustellen.
Die Destillation wurde von dem Chemiker Abu Musa Dschābir ibn Hayyān um 800 n. Chr. weiter verbessert. Auch der persische Wissenschaftler und Arzt Ar-Razi („Rhases“, 865–925) schrieb seine Kenntnisse in einer Reihe umfangreicher Schriften nieder. Sein wichtigstes Werk ist das Kitab sirr al-asrar, das „Buch des Geheimnisses der Geheimnisse“. Hier beschreibt er die Destillation des naft, des rohen Erdöls, und erläutert hierbei eine einfache Art des Crack-Verfahrens zum Zwecke der Gewinnung niedrig siedender Produkte wie Bitumen und des sogenannten Ziegelöles (oleum laterinum). Mit der Erfindung des Destillierhelms wurde auch die Destillation von Alkohol möglich.
Als um die erste Jahrtausendwende (1000 n. Chr.) die Schwefel- und Salpetersäure und vor allem der Trinkalkohol (Ethanol) entdeckt wurden, gewann die Destillation erheblich an Bedeutung. In der frühen Neuzeit wurde begonnen, die Destillation für medizinische Zwecke einzusetzen. So verfasste der Arzt Hieronymus Brunschwig 1512 Das Buch der wahren Kunst zu destillieren. Außerdem wurde die Destillation ein wichtiges Werkzeug in der Alchemie und später in der Spagyrik. Im 17. Jahrhundert begann man erneut mit der Süßwasserdestillation aus Meerwasser zur Meerwasserentsalzung. Die Destillation von Ethanol unterliegt in vielen Staaten Beschränkungen, Kontrollen und speziellen Steuern.
Prinzipien
Die oben beschriebene „einfache Destillation“ durch Erhitzen und Abkühlen beruht auf der Verdampfung und Kondensation flüchtiger Stoffe. Diese werden jedoch dadurch nicht oder nur unvollkommen getrennt. Man kann höchstens einzelne „Fraktionen“ mit unterschiedlicher Siedetemperatur auffangen.
Die Trennung von Gemischen verschiedener verdampfbarer und ineinander löslicher Stoffe kann oft durch wiederholte Destillation bewerkstelligt werden. In diesen Fällen beruht die Trennwirkung auf der unterschiedlichen Zusammensetzung der siedenden Flüssigkeit und des Dampfes. Eine notwendige, jedoch nicht ausreichende Bedingung hierfür sind unterschiedliche Siedepunkte der zu trennenden Komponenten. Die dafür entwickelten Techniken werden weiter unten aufgeführt. Diese Verfahren beruhen auf den unterschiedlich hohen Siedepunkten der beteiligten Flüssigkeiten, genauer gesagt auf ihrem unterschiedlichen Dampfdruck bei gleicher Temperatur. Dies sei an einem Gemisch aus zwei miteinander mischbaren flüssigen Komponenten („binäres Gemisch“) erläutert.
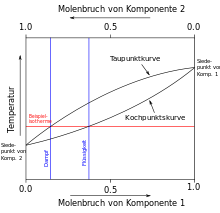
Wird wie in der Abbildung rechts eine Mischung aus den Komponenten 1 und 2 erhitzt, so steigt die Temperatur bis zum Erreichen der Siedekurve an. Die Zusammensetzung der Gasphase über der siedenden Flüssigkeit ist diejenige, welche die Taupunktkurve bei der gleichen Temperatur anzeigt (waagerechte Linie). Durch Kondensation erhält man eine Flüssigkeit, deren Zusammensetzung der der Gasphase entspricht, also einen erhöhten Anteil der niedriger siedenden Komponente 2 enthält (senkrechte Linie). Tatsächlich ist der Gehalt durch unvollständige Gleichgewichtseinstellung geringer. Außerdem verarmt die übrig bleibende Flüssigkeit (in der Technik: Destillationssumpf) mit der Zeit an der niedrigsiedenden Komponente, wodurch die waagerechte Linie nach oben verschoben wird.
Einfache Destillation
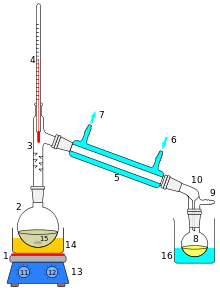
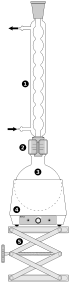
Bei der einfachen Destillation wird der Sumpf mithilfe einer geeigneten Wärmequelle (z.B. Heizhaube oder Heizbad) auf die gewünschte Temperatur erhitzt, bei der die Zielkomponente zu sieden beginnt. Ist diese erreicht, so steigt der Stoff in seinem gasförmigen Zustand auf, bis er im Kühler zu kondensieren beginnt. Im Labor wird dazu meist ein Liebigkühler verwendet. Anhand des Thermometers kann man die Kopftemperatur ablesen, die der verdampfte Stoff hat. Dies wird benötigt, um sicherzustellen, dass es sich um die gewünschte Komponente handelt, die man durch die Destillation aus dem Gemisch entfernen bzw. gewinnen möchte. Am Ende der Apparatur befindet sich der Auffangkolben.
Ein Beispiel für die Anwendung der einfachen Destillation ist die Abtrennung von Verunreinigungen einer Lösung durch Salze oder andere Partikel, die nicht durch Filtration entfernt werden können.
Mehrstufige Destillation und Rektifikation
Durch mehrfache erneute Destillation des Kondensates gelangt man im Siedediagramm auf einer Zickzacklinie immer näher an die Reinsubstanz 1 heran. In der Praxis erreicht man durch den Einbau einer Kolonne zwischen Destillationsblase und Destillenkopf schon durch einmalige Destillation eine deutlich erhöhte Trennleistung. Die Anzahl der für die gleiche Trennleistung benötigten Einzeldestillationen wird als „theoretische Bodenzahl“ bezeichnet, so genannt nach dem Verfahren der Erdöldestillation in sogenannten Glockenboden-Kolonnen. An der Oberfläche der Kolonne stellt sich durch Kondensation und Verdampfung das Gleichgewicht zwischen flüssiger und Gasphase ständig neu ein, wodurch nach oben hin der Anteil des niedrigsiedenden Bestandteils immer weiter ansteigt, während die höhersiedende Komponente in die Destillationsblase, den Sumpf, zurückfließt. Die Größe der Oberfläche der Kolonne, die im einfachsten Fall aus einem langen Glasrohr besteht, wird in verschiedene Varianten wie der Vigreux-Kolonne oder durch die Füllung mit Raschig-Ringen stark erhöht.
Falls die zu trennenden Stoffe ein Azeotrop bilden, so treffen sich Siede- und Taupunktkurve nicht erst bei den Reinsubstanzen. Eine destillative Trennung ist dann nur bis zu diesem Punkt möglich. Allerdings ist das azeotrope Mischungsverhältnis druckabhängig, so dass durch eine Vakuum- oder Überdruckdestillation doch eine weitere Trennung möglich ist. Das Azeotrop zwischen Ethanol und Wasser im Verhältnis ca. 25 : 1 (bei Umgebungsbedingungen) begründet die übliche Handelsmischung eines „96-prozentigen Alkohols“.
Die großtechnische Umsetzung der wiederholten, kontinuierlichen Destillation bezeichnet man auch als Rektifikation. Die einzelnen Destillationstufen finden in einem speziellen Behälter, Rektifikationskolonnen genannt, statt. Die Kolonne besteht aus mehreren Lagen von Böden, durch die der Dampf in den Kopf steigen und das Kondensat in den Sumpf fließen kann. Dabei können kontinuierlich Produkte abgezogen und Edukt nachgefüllt werden.
Fraktionierende Destillation
Ein aus mehreren Komponenten bestehendes Gemisch kann durch fraktionierende Destillation getrennt werden. Dabei wird der zum Auffangen des Destillates genutzte Behälter nach dem Abtrennen der am niedrigsten siedenden Fraktion ausgewechselt. Der Zeitpunkt zum Wechseln wird dabei durch eine Änderung der Temperatur im Destillationskopf angezeigt. Meist wird noch bis zum Erreichen des Siedepunkts der nächsten Komponente eine Zwischenfraktion abgetrennt, da im Übergangsbereich häufig ein Gemisch entsteht, und um Reste der vorherigen Fraktion aus dem Kühler zu entfernen. Liegen die Siedepunkte nahe beieinander, kann durch Zwischenschalten einer Kolonne das Volumen der unsauberen Zwischenfraktion klein gehalten werden.
- Hinweis
Die Begriffe „fraktionierende Destillation“ und „Rektifikation“ als Gegenstromdestillation, Rückflussdestillation, Kolonnendestillation werden häufig synonym verwendet. Im strengen Sinne bedeutet es, dass ein aus mehreren Komponenten bestehendes Gemisch durch Destillation und Fraktionierung getrennt werden kann. Dabei wird der zum Auffangen des Destillates genutzte Behälter nach dem Auffangen der am niedrigsten siedenden Fraktion ausgewechselt. Fraktionieren bedeutet dabei lediglich das Auffangen mehrerer Fraktionen.
Vakuumdestillation
Die Vakuumdestillation ist eine Destillation bei verringertem Gesamtdruck in der Destillationsanlage. Dadurch werden die Siedetemperaturen der einzelnen Komponenten gesenkt, was die Destillation von Stoffgemischen ermöglicht, deren im Sumpf verbleibenden Komponenten nicht ausreichend temperaturstabil sind. Bei höheren Temperaturen können im Sumpf oder im übrigen Edukt Katalysatorrückstände oder Nebenprodukte enthalten sein, die durch unerwünschte Reaktionen die Ausbeute senken.
Großtechnisch wird das „Sumpfprodukt“ der atmosphärischen Destillation bei der Erdölraffination anschließend noch einer Vakuumdestillation unterworfen. So sollen hauptsächlich die Grundöle zur Schmierölproduktion und sogenanntes Vakuumgasöl hergestellt werden. Dies dient weiterhin als wertvolles Edukt für einen Cat Cracker oder einen Hydrocracker.
Überdruckdestillation
Bei der Überdruckdestillation wird die Anlage mit Überdruck gefahren, um die Siedepunkte weiter auseinander zu schieben. Der Anwendungsbereich liegt bei Stoffen mit sehr niedrigen Siedepunkten, die eng beieinander liegen, wie bei der Luftverflüssigung.
Auch bei Pflanzenmaterial mit schwer destillierbaren Ölen wird zuweilen die Überdruckdestillation mit überhitztem Wasserdampf angewandt. Hierbei ist das Öl-Wasser-Verhältnis im Destillat günstiger als bei Normaldruck.
Kugelrohrdestillation
Destillationen im Kugelrohr werden im Labor mit kleinen Substanzmengen durchgeführt. Näheres wird unter diesem Lemma beschrieben.
Schleppdestillation
Hierbei wird mit einem Stoffzusatz destilliert, der das Produkt „mitschleppt“. Bekannteste Variante dieser Destillationsart ist die Wasserdampfdestillation. Wenn eine Vakuumdestillation nicht optimal durchzuführen ist, wird diese angewandt, um wärmeempfindliche Substanzen mit geringem Dampfdruck zu destillieren. Beispiele sind die Extraktion von ätherischen Ölen aus Pflanzen oder die Anwendung bei der Reinigung substituierter Aromaten.
Azeotrope Destillation
Hierbei wird eine Komponente zugegeben, die mit dem abzutrennenden Stoff ein Azeotrop bildet. Beispielsweise kann bei einer sauer katalysierten Veresterung das entstehende Wasser als Azeotrop mit Toluol quantitativ entfernt werden, wodurch die Reaktion erst vollständig abläuft. Idealerweise bildet sich ein Heteroazeotrop, das beim Kondensieren wiederum in zwei Phasen zerfällt, was eine Rückführung des Lösungsmittels erlaubt.
Kurzwegdestillation
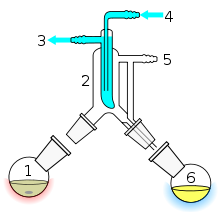
Als Kurzwegdestillation (KWD) bezeichnet man eine Destillation, die im Feinvakuumbereich, also im Druckbereich zwischen 1 und 0,001 mbar durchgeführt wird und bei der die Gasphase nur einen sehr kurzen Weg zwischen der Vorlage und dem Kondensator zurückzulegen hat. Sie wird auch als Molekulardestillation bezeichnet und gehört zu den schonendsten thermischen Trennverfahren. Aufgrund des geringen Arbeitsdrucks erfolgt die Destillation schon bei relativ niedrigen Temperaturen. Im Vergleich zu anderen Destillationsverfahren können somit thermisch empfindliche Produkte wie Tocopherole, Fettsäureester, Monoglyceride, Prepolymere, Epoxidharze und Pharmawirkstoffe sehr schonend getrennt werden. Geeignet ist die Methode auch für schwer verdampfbare Moleküle, wie langkettige Kohlenwasserstoffe aus den Rückständen der Mineralölindustrie, die unter Feinvakuum abdestilliert werden. Eine modifizierte Variante ist die Kugelrohrdestillation. In der Industrie sind plattenwärmetauscherähnliche Apparate im Einsatz, bei denen der Abstand zwischen Verdampfer und Kondensator nur wenige Millimeter beträgt.
Reaktivdestillation
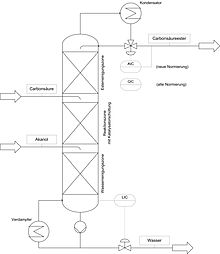
Bei der Reaktivdestillation wird die (mehrstufige) Destillation mit einer chemischen Reaktion kombiniert. Durch die Kombination beider Mechanismen können Vorteile im Vergleich zu einfachen, seriellen Reaktions-Destillations-Verfahren erzielt werden. Reaktivdestillation eignet sich vor allem für „gleichgewichtslimitierte“ Reaktionen. Durch das ständige Entfernen eines Reaktionspartners wird das Gleichgewicht immer wieder neu eingestellt und auf diese Weise ein vollständiger Umsatz ermöglicht. Andererseits können durch die Reaktion auftretende Azeotrope gebrochen werden. Bei exothermen Reaktion wird dabei die auftretende Wärme zur Stofftrennung ausgenutzt. Die optimalen Betriebsbedingungen und hierbei vor allem der optimale Temperaturbereich für Reaktion und Stofftrennung können diese Methode verhindern.
Die auftretende chemische Reaktion kann sowohl homogen als auch heterogen katalysiert werden. Bei der Verwendung eines homogenen Katalysators ist eine weitere Trennstufe zur Abtrennung des Katalysators notwendig. Bei der heterogen katalysierten Reaktivdestillation wird der Katalysator häufig in Form von reaktiven Packungen in der Destillationskolonne eingebaut. Dabei handelt es sich oftmals um Trennpackungen, in die der meist kugelförmige Katalysator in Metallsäckchen integriert ist. Trotz intensiver Forschungen in den letzten Jahrzehnten findet die Reaktivdestillation in der Industrie nur relativ selten Verwendung. Wichtig ist sie allerdings bei der Kaliumproduktion.
Zonendestillation
Die Zonendestillation ist ein Destillationsprozess in einem Container gestreckter Form mit partieller Verschmelzung des raffinierten Stoffes in einer sich bewegenden flüssigen Zone und mit einer Kondensation des Dampfes in die feste Phase im Zuge des Ausgangs des Kondensats zum kalten Gebiet. Der Prozess ist theoretisch bearbeitet.
Bei der Bewegung des Zonenerhitzers den Container entlang von oben nach unten lässt sich ein festes Kondensat im Container mit der gleichmäßigen Verteilung der Beimischungen formen und der reinste Teil des Kondensats kann als Produkt ausgegrenzt werden. Der Prozess kann mehrmals wiederholt werden, wofür das früher erhaltene Kondensat (ohne Umwälzung) in den unteren Teil des Containers an den Ort des raffinierten Stoffes versetzt werden soll. Die ungleichmäßige Verteilung der Beimischungen im Kondensat (d.h. die Reinigungswirkung) steigt mit der Anzahl der Wiederholungen des Prozesses.
Die Zonendestillation ist ein destillatives Analogon der Zonenumkristallisation. Die Verteilung der Beimischungen im Kondensat wird durch bekannte Gleichungen der Zonenumkristallisation mit verschiedener Anzahl der Durchläufe der Zone beschrieben – bei der Ersetzung des Verteilungskoeffizienten k für die Kristallisation durch den Separationskoeffizienten α für die Destillation.


© biancahoegel.de
Datum der letzten Änderung: Jena, den: 17.05. 2026