Chromatographie

Chromatographie, Chromatografie (griechisch, χρῶμα chroma „Farbe“ und γράφειν graphein „schreiben“, zu deutsch Farbenschreiben) wird in der Chemie ein Verfahren genannt, das die Auftrennung eines Stoffgemisches durch unterschiedliche Verteilung seiner Einzelbestandteile zwischen einer stationären und einer mobilen Phase erlaubt. Dieses Prinzip wurde erstmals 1901 von dem russischen Botaniker Michail Semjonowitsch Zwet beschrieben, 1903 wurde es zum ersten Mal öffentlich gedruckt beschrieben, 1906 benutzte er erstmals den Begriff „Chromatographie“. Er untersuchte gefärbte pflanzliche Extrakte, zum Beispiel aus Blattmaterial, und konnte daraus durch Chromatographie verschiedene Farbstoffe isolieren. Anwendung findet diese Methode zum einen in der Produktion zur Reinigung von Substanzen (= präparative Chromatographie), zum anderen in der chemischen Analytik, um Stoffgemische in möglichst einheitliche Inhaltsstoffe zwecks Identifizierung oder mengenmäßiger Bestimmung aufzutrennen. Die Chromatographie wird in der organischen Chemie, der Pharmazie, der Biochemie, der Biotechnologie, der Mikrobiologie, der Lebensmittelchemie, der Umweltchemie und auch in der anorganischen Chemie angewendet.
Prinzip der Chromatographie
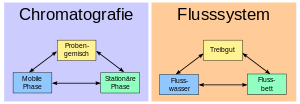
Die Chromatographie lässt sich am einfachsten durch einen Vergleich erklären:
Ein reißender Fluss kann einiges an Treibgut mit sich führen. Die Geschwindigkeit, mit der das Treibgut weiterbewegt wird, hängt ab
- von der Art des Treibguts (Sandkörner werden schneller als Kieselsteine transportiert),
- von der Beschaffenheit des Flussbetts (raue Oberflächen erhöhen die Reibung des Treibguts und verringern somit die Geschwindigkeit des Abtransports)
- von der Strömungsgeschwindigkeit.
In der Chromatographie werden unterschiedliche Substanzen (= Treibgut) in der so genannten mobilen Phase (= Wasser) auf einer stationären Phase (= Flussbett) befördert. Aufgrund der Wechselwirkungen (siehe die Einteilung unter Trennprinzipien) zwischen der Probe, der stationären Phase und der mobilen Phase werden einzelne Substanzen unterschiedlich schnell weitertransportiert und somit voneinander getrennt: Ein Gemisch aus Sand, sehr kleinen und etwas größeren Kieselsteinen wird an einer Stelle des Flusses eingebracht; nach beispielsweise hundert Metern kommt zuerst der gesamte Sand an (verteilt auf ein paar Meter) und nach einer gewissen Wartezeit kommen alle kleineren Kieselsteine und noch viel später die größeren, jeweils auf eine gewisse Strecke auseinandergezogen.
Dieser Vergleich eignet sich für einen ersten Einstieg. Tatsächlich erinnert der Prozess (bei der Chromatographie) eher an einen „digitalen“ Prozess (engl. „stop and go“). Die Probemoleküle werden entweder mit der mobilen Phase mitgenommen (mit der Geschwindigkeit der mobilen Phase – Analogie wäre ein Floß, das passiv in einem Strom mitgeführt wird) oder sie haften an der stationären Phase (Geschwindigkeit gleich Null). Zwischen diesen beiden Möglichkeiten wechseln sie sehr rasch hin und her (aufgrund der Wärmebewegung erhalten sie ständig Stöße). Der Vergleich mit dem Flussbett könnte auch zu einem weiteren Missverständnis führen: die Verzögerungen, die die verschiedenen Probemoleküle auf dem Weg durch das chromatographische System erleiden, haben nichts mit Reibungsphänomenen zu tun. Basis für das Verständnis sind Unterschiede in der Verteilung (der verschiedenen Molekülsorten A, B, C usw.). Sie entsprechen Unterschieden im Zeitanteil (den die einzelnen Moleküle vom Typ A, B, C usw. im Mittel in der mobilen Phase verbringen). Die Chromatographie schafft es, diese Unterschiede in Geschwindigkeitsunterschiede zu verwandeln und damit für eine Trennung gut nutzbar zu machen. Das könnte man auch als den „Trick“ oder als das Prinzip der Chromatographie bezeichnen. Ansonsten wären diese oft recht kleinen Unterschiede kaum zu nutzen, weder für Trenn- und Reinigungsprozesse, noch für Analysen.
Anhand eines Beispiels ist es leichter zu verstehen: Wenn sich 45 % der A-Moleküle in der mobilen Phase aufhalten (im Mittel), kommt es auf Grund des dynamischen Gleichgewichtes dazu, dass die individuellen A-Moleküle auch 45 % der Zeit in der mobilen Phase verbringen (im Mittel). Daher wird ihre Geschwindigkeit 45 % der Geschwindigkeit der mobilen Phase betragen (im Mittel). Für gute Ergebnisse bei der Chromatographie ist es entscheidend, dass der Stoffaustausch zwischen den beiden Phasen sehr rasch erfolgt, das heißt die einzelnen Probemoleküle sollen sehr oft zwischen den beiden Phasen hin und her wechseln (Diffusionsprozesse, Wärmebewegung). Eine Voraussetzung dafür ist, dass die Wege, welche die Moleküle von der stationären Phase zur mobilen Phase zurückzulegen haben, sehr kurz sind. Falls die stationäre Phase ein Pulver enthält, sollte die Korngröße dieses Pulvers sehr klein sein (zum Beispiel nur wenige Mikrometer). Aus bestimmten Gründen sollten die Pulverkörner auch möglichst einheitlich geformt sein und eine möglichst einheitliche Größe aufweisen (enge Korngrößenverteilung).
Prozess
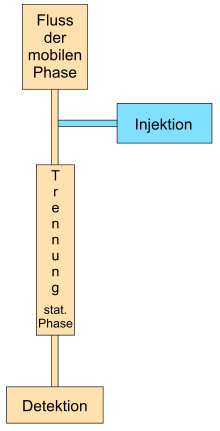
Für die Chromatographie sind die Herstellung des Flusses der mobilen Phase, die Injektion der zu trennenden Probe, die eigentliche Trennung und die Detektion nötig. Das Fließen der mobilen Phase wird entweder mittels Druck (hydraulischen Pumpe, Gasdruck), Kapillarkraft oder durch Anlegen einer elektrischen Spannung erreicht.
Die Injektion (= Einbringen des Substanzgemisches in das chromatographische System) erfolgt entweder, bevor der Fluss der mobilen Phase hergestellt wird (z.B. Dünnschichtchromatographie) oder während die mobile Phase bereits fließt. Bei einer großen Anzahl von Proben werden bei automatisierbaren Chromatographiearten sogenannte Autosampler (zusammen mit eigenen Datenerfassungssystemen) eingesetzt, die vollautomatisch die Proben injizieren.
Anschließend erfolgt die eigentliche Auftrennung des Substanzgemisches auf der Trennstrecke. Ohne Detektion (= sichtbar machen, wann eine Substanz einen bestimmten Teil des Chromatographiesystems passiert oder wo eine Substanz nach dem Beenden des Prozesses zum Liegen kommt) ist eine Chromatographie nicht denkbar. Für jede Chromatographieart werden verschiedene Detektionssysteme eingesetzt, indem entweder physikalische Eigenschaften (Absorption von Licht, Fluoreszenz, Lichtstreuung, Wärmeleitfähigkeit.) der Substanzen ausgenutzt werden oder durch chemische Reaktionen ein Signal erhalten wird. Mittels chemischer Reaktionen wird z.B. eine Färbung bei der planaren Chromatographie erreicht (z.B. Aminosäuren mittels Ninhydrin) oder Reaktionen vor dem Auftrennen (Vorsäulenderivatisierung) oder nach dem Auftrennen (Nachsäulenderivatisierung) bei der Säulenchromatographie durchgeführt.
Bei der präparativen Chromatographie wird anschließend noch ein Fraktionensammler zum Auffangen der aufgetrennten Substanz benötigt.
Bauartbedingt handelt es sich bei chromatographischen Aufreinigungsverfahren immer um Batch-Verfahren. Dies bedeutet, dass immer nur eine bestimmte Menge an Substanz aufgetragen und getrennt werden kann, bevor mit der nächsten Menge fortgefahren werden kann. Dies ist insbesondere bei der Aufarbeitung großer Mengen problematisch, so dass einige Verfahren entwickelt wurden, um Chromatographie kontinuierlich betreiben zu können: Annulare Chromatographie, TMB (True Moving Bed) Chromatographie und SMB-(Simulated Moving Bed) Chromatographie.
Begrifflichkeiten und Prinzip
Stationäre Phase
Phase, die mit den einzelnen Substanzen des Substanzgemisches Wechselwirkungen eingeht und sich nicht bewegt. Der Aufenthalt der Analyten bei ihrer Retention wechselt zwischen mobiler und stationärer Phase (random walk) und verursacht die substanzcharakteristische Retentionszeit. Die stationäre Phase besteht in der Gaschromatographie aus einer Flüssigkeit (Trennflüssigkeit) oder einem Gel, mit welchem die Innenseite der Kapillare beschichtet ist. Bei der Flüssigchromatographie ist die stationäre Phase normalerweise fest, kann aber auch eine Flüssigkeit sein, die mit der mobilen Phase nicht mischbar ist und den pulverförmigen Träger benetzt. Schließlich kann die stationäre Phase auch aus chemisch an den Träger gebundenen Molekülen bestehen.
Mobile Phase
Phase, in die das Substanzgemisch zu Beginn des Trennsystems eingebracht und die bewegt wird. Bei der Flüssigchromatographie ist die mobile Phase flüssig. Bei der Gaschromatographie kommen Trägergase wie Wasserstoff, Helium oder Stickstoff zum Einsatz, in der Dünnschichtchromatographie spricht man vom Fließmittel. Mobile Phasen unterscheiden sich in ihrer Elutionsfähigkeit („Stärke“ s.u. „Elutrope Reihe“), dies bedingt unterschiedliche Retentionszeiten und oft auch unterschiedliche Selektivitäten.
Retention
Unter Retention versteht man den verzögerten Durchfluss einzelner Substanzen des Substanzgemisches der mobilen Phase durch Wechselwirkung mit der stationären Phase.
Die Retention einer Substanz durch die stationäre Phase wird im Wesentlichen durch drei Aspekte bestimmt:
- Stärke der Wechselwirkung der Substanz mit der stationären Phase („Neigung in der stationären Phase zu bleiben“)
- Siedepunkt der Substanz („Neigung in der mobilen Phase zu bleiben“)
- Diffusionseigenschaften der Substanz („Beweglichkeit in der stationären und mobilen Phase“)
In vielen Fällen wird gezielt eine spezielle Wechselwirkung des zu analysierenden Stoffes mit der stationären Phase genutzt, um Substanzen zu trennen. Die Stärke der Wechselwirkungen zwischen den Probenkomponenten und der stationären Phase wird sowohl von deren Struktur als auch von deren funktionellen Gruppen bestimmt. Dabei treten bei unpolaren Substanzen ausschließlich Dispersionswechselwirkungen (Van-der-Waals-Bindung) auf, während polare Trennphasen auch polare Wechselwirkungen eingehen können, etwa Wasserstoffbrückenbindungen oder Donator-Akzeptor-Bindungen. Letztere trennen nach dem Prinzip: Gegensätze ziehen sich an. Das bedeutet, dass Trennphasen, die etwa Wasserstoff zur Wasserstoffbrückenbindung aufzunehmen in der Lage sind Substanzen trennen, die Wasserstoff zur Brückenbindung bereitstellen können (etwa Alkohole). Auch können zum Beispiel Enantiomere, welche sich in ihren Siedepunkten nicht unterscheiden und somit gleiche Retentionszeiten aufweisen würden, durch ihre verschieden starken Wechselwirkungen mit speziellen Derivaten von Cyclodextrinen aufgetrennt werden.
Retentionszeit
Zeit, die die Moleküle eines reinen Stoffes zum Durchwandern der Säule benötigen (von der Injektion bis zur Detektion).
Im Gegensatz zu den Trägergasen zeigen die meisten chemischen Stoffe eine Wechselwirkung mit der stationären Phase, d.h., sie halten sich für eine gewisse Zeit in der stationären Phase auf. Ihre Aufenthaltsdauer in der stationären Phase addiert sich zur Aufenthaltsdauer in der mobilen Phase (Totzeit), sie benötigen also insgesamt länger, um die ganze GC-Säule zu passieren. Ursprünglich leitet sich der Begriff Retention (Zurückhaltung) davon ab, dass die stationäre Phase den Analyten für eine gewisse Zeit zurückhält. Heutzutage wird der Begriff Retentionszeit allerdings vereinfacht verwendet für die Zeit, die der Analyt zum Passieren der Säule benötigt und dies schließt also die Totzeit mit ein. Daher werden die Begriffe folgendermaßen definiert:
- Retentionszeit (tR): Ist die Gesamtzeit, die ein Analyt für das Passieren der Säule benötigt. Dies entspricht der Zeit zwischen Injektion und Detektion des Analyten.
- Totzeit (t0): Ist die Zeit, in der sich ein Analyt ohne Wechselwirkung mit der stationären Phase in der mobilen Phase aufhält; entspricht also der Zeit, die die mobile Phase zum Durchlaufen der Säule benötigt.
- Nettoretentionszeit oder reduzierte Retentionszeit (tN): Ist die Differenz aus Retentionszeit und Totzeit. Sie entspricht also der Zeit, in der sich ein Analyt in der stationären Phase aufhält. tN = tR - t0
Durchflusszeit (Totzeit)
Die Durchflusszeit (auch „Totzeit“) gibt die Zeit an, die die mobile Phase oder eine nicht zurückgehaltene Substanz benötigt, um die Chromatographie-Apparatur von der Injektion über die Säule bis zum Detektor zu durchwandern (s.a. Durchflussvolumen). Die Durchflusszeit kann bestimmt werden, indem eine nicht zurückgehaltene Substanz („Inertsubstanz“) injiziert wird. Diese Substanz geht nur in sehr geringem Maße Wechselwirkungen mit der stationären Phase ein. Sie durchläuft daher die Apparatur in derselben Zeit wie die mobile Phase. Die Durchflusszeit ist dann identisch mit der Zeit zu der der Peak im Detektor erscheint.
Durchflussvolumen
Das Durchflussvolumen kann direkt aus der Durchflusszeit abgeleitet werden. Es ergibt sich aus der einfachen Formel Durchflussvolumen = Fluss der mobilen Phase · Durchflusszeit. Das Durchflussvolumen ist für zahlreiche Berechnungen in der Hochleistungsflüssigkeitschromatographie (HPLC) sehr wichtig, z.B. für den Methodentransfer zwischen Säulen mit unterschiedlichem Volumen.
Elution
Elution (von lat. eluere „auswaschen“) ist das Herauslösen oder Verdrängen von adsorbierten Stoffen aus festen oder mit Flüssigkeit getränkten Adsorbentien und Ionenaustauschern durch kontinuierliche Zugabe eines Lösungsmittels (Elutionsmittel = mobile Phase). Die aus der Trennsäule fließende Lösung wird Eluat genannt.
Eine besondere Bedeutung hat dieser Prozess in der Festphasenextraktion.
Eluotrope Reihe
Anordnung der als mobile Phase üblichen Lösungsmittel nach ihrer Elutionskraft bei einer Referenzsubstanz (i. d. R. Kieselgel oder Aluminiumoxid).
Bluten
Als Bluten wird ein Effekt bei Chromatographie-Säulen bezeichnet, bei dem die Säule geringe Anteile ihrer Matrix verliert. Man spricht auch von Säulenbluten. Ursache für verstärktes Säulenbluten können in der Gaschromatographie eine übermäßige thermische Belastung der Säule und in der Hochleistungsflüssigkeitschromatographie (HPLC) die Verwendung zum Beispiel ungeeigneter pH-Werte des Elutionsmittels oder der Eluenten (zu stark sauer oder alkalisch) sein. Säulenbluten findet in geringem Ausmaß auch im laufenden Betrieb statt und stellt dort normalerweise kein Problem dar. Durch das Säulenbluten ergibt sich, unter anderem, die Alterung von Trennsäulen. Starkes Säulenbluten bewirkt jedoch ein starkes Signalrauschen und einen hohen Hintergrundwert bei der Detektion oder nachgeschalteten Analyseverfahren, wie einem Massenspektrometer.
Säule
In der Chromatographie versteht man unter einer Säule, oder Trennsäule, eine hohle Röhre mit einem Durchmesser von wenigen Mikrometern bis zu mehreren Metern. Auch die Länge variiert von wenigen Zentimetern bis zu 150 Metern. In dieser Röhre ist entweder nur die Innenwand beschichtet (Kapillarsäule), oder die Säule ist mit der stationären Phase befüllt (gepackte Säule). Von der Säule im Bauwesen unterscheidet sie sich insofern, als sie weder gerade noch senkrecht sein muss, sondern auch wie ein Schlauch aufgerollt sein kann.
Reversed-Phase-Mechanismus
Bei der Adsorptionschromatographie gibt es zwei Möglichkeiten ein Substanzgemisch zu trennen:
- Normale Phase: polare stationäre Phase (wie Kieselgel, Aluminiumoxid), unpolare bis mittelpolare mobile Phase (wie Kohlenwasserstoffe, Dioxan, Essigsäureethylester …) oder
- Reversed Phase (Umkehrphase): unpolare stationäre Phase (wie modifiziertes Kieselgel) und polare mobile Phase (wie gepuffertes Wasser).
Im ersten Fall werden lipophile Stoffe leicht, polare schwer eluiert, im Umkehrfall polare leicht eluiert („similia similibus solvuntur“).
In der Hochleistungsflüssigkeitschromatographie wird oft eine Gradienteneluierung angewandt, wobei die Zusammensetzung des Lösungsmittels langsam geändert wird (z.B. von 80 % auf 20 % Wasseranteil). So treten Alkane sehr spät und Aminosäuren sehr früh aus der Säule aus und man kann diese Fraktionen herausschneiden.
Einteilung nach dem Trennprinzip
Das grundlegende Prinzip aller chromatographischen Verfahren ist die oft wiederholte Einstellung eines Gleichgewichtes zwischen einer ruhenden Phase und einer bewegten Phase. Das Gleichgewicht kann sich auf Grund verschiedener physikalisch-chemischer Effekte ausbilden.
- Adsorptionschromatographie – Hier kommt es zu einer Trennung der verschiedenen Komponenten auf Grund der unterschiedlich starken adsorptiven Bindungen zur ruhenden Phase. Die bewegte Phase kann ein mehr oder weniger polares Lösungsmittel oder bei gasförmigen Stoffen ein Trägergas sein. Im Fall der Flüssigchromatographie stellt man sich einen Wettkampf zwischen den diversen Probemolekülen mit den Molekülen der mobilen Phase (Fließmittel, Laufmittel) um die Haftstellen auf der (großen) Oberfläche der stationären Phase vor.
- Verteilungschromatographie – Ähnlich dem Extraktionsverfahren wird hier die unterschiedliche Löslichkeit der zu trennenden Komponenten ausgenutzt. Bei der Chromatographie bleibt aber das Lösungsmittel als ruhende Phase auf einem Trägermaterial haften. Die bewegte Phase kann wieder eine Lösung oder ein Trägergas sein.
- Ionenaustauschchromatographie
– Die bewegte Phase ist hier meist eine Lösung der zu trennenden Ionen. Die
ruhende Phase ist ein fester Ionenaustauscher.
Ionenaustauscher bilden zwischen den verschiedenen Ionen der bewegten Phase
unterschiedlich stabile Bindungen aus.
- Chelatbildner-Chromatographie ist eine spezielle Form der Kationenaustausch-Chromatographie. Die ruhende Phase bindet selektiv polyvalente Kationen über komplexbildende, funktionelle Gruppen. Die Selektivität für Schwermetall-Ionen gegenüber von Alkali- und Erdalkali-Ionen ist hoch.
- Siebwirkung – Bei der ruhenden Phase benutzt man Stoffe, die die
Komponenten anhand ihrer Größe trennen. Im Wesentlichen unterscheidet man hier
zwischen drei Verfahren.
- Molekularsieb-Chromatographie
- Gel-Permeations-Chromatographie (Gelfiltration)
- Ausschluss-Chromatographie
- Bei einem Sieb haben Teilchen „Vorteile“, die fein genug sind, um die Poren des Siebes zu durchdringen. Bei den entsprechenden Chromatographieverfahren ist es genau umgekehrt. Genügend feine Teilchen sind in der Lage, sich in die Hohlräume der stationären Phase zu „verirren“, sind daher langsamer unterwegs als Moleküle, die auf Grund ihrer Größe aus diesen Hohlräumen (mehr oder weniger oder zur Gänze) ausgeschlossen werden. Bei genügender Größe wandern sie unverzögert, da sie sich nur im bewegten Flüssigkeitsstrom aufhalten und nie im unbewegten Teil der Flüssigkeit (in den Hohlräumen – z.B. des Gels).
- Affinitätschromatographie
– Als stationäre Phase wird eine für jeden Analyten spezifische chemische
Verbindung eingesetzt, die auf Grund nichtkovalenter Kräfte eine Trennung
bewirkt. Es handelt sich hierbei um eine hochselektive Methode.
- IMAC (Immobilized Metal Ion Affinity Chromatography) – Hierbei werden über Komplexbindungen Metallionen wie Fe, Co, Ga u.a. an eine Matrix gebunden. Die Trennung wird über die unterschiedlichen Wechselwirkungen zwischen dem Analyten und den Metallionen erreicht. Besonders bei der Aufreinigung von Proteinen durch Phosphorylierung hat sich diese Methode etabliert. Dabei werden als Metallionen vornehmlich Fe und Ga genutzt. Als Konkurrenzverfahren hat sich dort seit einiger Zeit die Anreicherung über Titandioxid-Säulen bewährt. Für die Aufreinigung von poly-Histidin-markierten Proteinen sind ebenfalls verschiedene IMAC-Verfahren vorhanden. Hierbei werden vor allem Ni und Co Ionen für die Anreicherung genutzt.
- Thiol-Disulfid-Austausch-Chromatographie verwendet auf der ruhenden Phase fest gebundene Thiol-Gruppen um die Thiol-Gruppen auf den Molekülen in der bewegten Phase als kovalente Disulfide reversibel zu binden. An diesen Bindungen beteiligen sich vor allem jene Thiol-Gruppen, die an der Außenseite der Proteinmoleküle liegen.
- Chirale Chromatographie – Zur Trennung von chiralen Molekülen. Die stationäre Phase enthält ein Enantiomer, das mit den beiden Enantiomeren des Racemats eine unterschiedlich starke diastereomere Wechselwirkung eingeht. Die beiden Enantiomere werden unterschiedlich stark retardiert.
Einteilung nach den verwendeten Phasen
Aufgrund der mobilen Phasen kann man die Chromatographie in drei Gebiete unterteilen, welche sich nach den Trägern der stationären Phasen oder der Dichte einteilen lassen
- Flüssigchromatographie
(engl. liquid Chromatography, LC)
- Planare Chromatographie
- Papierchromatographie – Als feste Phase wird Papier verwendet, das entweder liegt oder (meist) senkrecht in einem Glasbehälter steht. Wie auch bei der Dünnschichtchromatographie wird die mobile Phase auf Grund der Kapillarkräfte bewegt.
- Dünnschichtchromatographie – Als feste Phase werden z.B. Silikapartikel in einer feinen Schicht auf einer flexiblen Trägerfolie aus Aluminium oder Plastik oder einer Glasplatte aufgetragen. Eine Variante ist die zirkuläre DC, mit einer rotierenden, beschichteten Kreisscheibe (speziell für präparative Zwecke geeignet).
- Säulenchromatographie
- Niederdruckchromatographie – Die hier verwendeten Säulen weisen Durchmesser von einem bis mehreren Zentimetern auf. Diese Form der Flüssigchromatographie wird vor allem für präparative Trennungen eingesetzt.
- Hochleistungsflüssigkeitschromatographie (unrichtig, jedoch sehr gebräuchlich ist auch der Ausdruck Hochdruckchromatographie; engl. HPLC High Performance (Pressure) Liquid Chromatography) – Sie stellt die heute am weitesten verbreitete in der Analytik eingesetzte Trennmethode dar, die eigentlich unkorrekte (veraltete) Bezeichnung High Pressure Liquid Chromatography bezieht sich auf die Drücke, die diese Methode von der Niederdruck- oder anderen Chromatographiearten unterscheidet. Immerhin werden hier bis über 1000 bar bei einer Flussrate der mobilen Phase bis zu 5 ml/min erzeugt, die jedoch mit der Trennleistung nicht zu tun haben, sondern nur zur Fortbewegung des Eluentengemischs in der Säule dienen.
- Elektrochromatographie – In diesem Fall wird die Mobile Phase durch Anlegen einer Spannung bewegt. Diese Methode befindet sich noch im Entwicklungsstadium und wird im Routinebetrieb nicht angewendet. Nicht zu verwechseln mit Elektrophorese.
- Membranchromatographie
- Hierbei wird statt einer mit chromatographischer Matrix gefüllten Säule eine ein- oder mehrlagige Membran als feste Phase in einem entsprechenden Gehäuse eingesetzt. Die mobile Phase wird bei niedrigen Drücken bis zu 6 bar und bei etwa 20-fach höheren Flussraten als in der Säulenchromatographie üblich durch die Membran gepumpt.
- Planare Chromatographie
- Gaschromatographie
- Gepackte Säulen – Das Innere einer Säule (lange Röhre) ist mit einem feinkörnigen Material gefüllt. In der Regel besteht dabei die stationäre Phase aus einem dünnen Film einer weitgehend inerten und hochsiedenden Flüssigkeit, der die Pulverkörner überzieht.
- Kapillarsäulen – Nur die Säulenwand ist mit einer dünnen Schicht
aus stationärer Phase bedeckt.
- Flüssige stationäre Phase
- Feste stationäre Phase
- Überkritische Fluidchromatographie (engl. SFC supercritical fluid chromatography) – Als mobile Phase wird eine Substanz in ihrer überkritischen Phase (Zustand zwischen Gas und Flüssigkeit) eingesetzt. Hierbei handelt es sich meist um Kohlendioxid. Bei dieser Methode werden nur Säulen als Träger von stationären Phasen eingesetzt.
Kenngrößen der Chromatographie
- Säulenlänge

 nennt man die lineare Durchflussgeschwindigkeit der mobilen Phase durch
die Säule, sie ist definiert als:
nennt man die lineare Durchflussgeschwindigkeit der mobilen Phase durch
die Säule, sie ist definiert als:

- der Retentionsfaktor
 ist definiert durch
ist definiert durch

- Der Selektivitätskoeffizient α gibt die Güte der Trennung zweier
Substanzen an. Er beruht auf den Retentionszeiten
 der Komponenten in der Säule. Die Retentionszeit ist die Zeit, die die
betrachtete Komponente zum Durchqueren der Säule braucht und wird am
Peakmaximum abgetragen:
der Komponenten in der Säule. Die Retentionszeit ist die Zeit, die die
betrachtete Komponente zum Durchqueren der Säule braucht und wird am
Peakmaximum abgetragen:

 ,
die chromatographische Auflösung (Resolution) zweier Peaks
errechnet sich aus:
,
die chromatographische Auflösung (Resolution) zweier Peaks
errechnet sich aus:
 oder
oder
- Der Faktor 1,18 kommt durch das Verhältnis der Halbwertsbreite zur Basisbreite einer Gaußschen Glockenkurve ((2 · ln 2)0,5) zustande.
 ,
die Trennstufenzahl oder Bodenzahl beschreibt die Anzahl der
Gleichgewichtseinstellungen, der zu trennenden Substanz zwischen stationärer
und mobiler Phase, in der Säule. Je größer N, desto mehr
Gleichgewichtseinstellungen können in einer bestimmten Länge erfolgen, woraus
eine bessere Trennleistung der Säule resultiert. N wird berechnet mit Hilfe
der Formel:
,
die Trennstufenzahl oder Bodenzahl beschreibt die Anzahl der
Gleichgewichtseinstellungen, der zu trennenden Substanz zwischen stationärer
und mobiler Phase, in der Säule. Je größer N, desto mehr
Gleichgewichtseinstellungen können in einer bestimmten Länge erfolgen, woraus
eine bessere Trennleistung der Säule resultiert. N wird berechnet mit Hilfe
der Formel:
 oder
oder
 :
Basislinienbreite
:
Basislinienbreite
 :
„Full Width at Half Maximum“ Fwhm
:
„Full Width at Half Maximum“ Fwhm
 :
Peakkapazität; Gibt an, wie viele Peaks innerhalb eines Intervalls zwischen
:
Peakkapazität; Gibt an, wie viele Peaks innerhalb eines Intervalls zwischen
 und dem k-Wert eines bestimmten Peaks theoretisch mit einer Auflösung von
R=1,5 (Basislinientrennung) voneinander getrennt werden können.
und dem k-Wert eines bestimmten Peaks theoretisch mit einer Auflösung von
R=1,5 (Basislinientrennung) voneinander getrennt werden können.

 bezeichnet die Trennstufenhöhe (oder theoretische Bodenhöhe)
eines theoretischen Bodens (HETP - ‚Höhenäquivalent eines theoretischen
Bodens‘, englisch
‚height equivalent to a
theoretical plate‘) und ist das Verhältnis zwischen Säulenlänge
und Bodenzahl :
bezeichnet die Trennstufenhöhe (oder theoretische Bodenhöhe)
eines theoretischen Bodens (HETP - ‚Höhenäquivalent eines theoretischen
Bodens‘, englisch
‚height equivalent to a
theoretical plate‘) und ist das Verhältnis zwischen Säulenlänge
und Bodenzahl :

- Praktische Werte liegen im Bereich von 0,1 bis 0,5 mm.
Trennstufenhöhe H
Die Trennstufenhöhe einer chromatographischen Säule ist ein Maß für die Trennleistung der Säule. Als Trennstufe kann man sich den gedachten Abschnitt der Trennsäule, auf dem sich das chromatographische Gleichgewicht einmal einstellt, vorstellen. Je mehr solche Gleichgewichtseinstellungen „auf der Säule Platz haben“, umso geringer ist die Trennstufenhöhe und umso höher ist die Trennleistung der Säule. Zur Erlangung einer niedrigen Trennstufenhöhe sind unter analytischen Bedingungen folgende Voraussetzungen nötig:
- Es wird eine rasche Gleichgewichtseinstellung der Adsorption oder Verteilung erwartet. Daher sollte der Teilchendurchmesser so klein wie möglich sein.
- Konstante Temperatur in der gesamten Säule. Dazu kann ein Säulenthermostat benutzt werden.
- Konstante Fließgeschwindigkeit: Hierfür wird eine Kolbenpumpe mit bis zu 400 bar verwendet.
- Linearer Adsorptionsbereich: Die stationäre Phase sollte im Verlauf der Chromatographie nicht überladen werden.
- Vernachlässigbare Diffusion wäre wünschenswert, ist experimentell aber leider nicht erreichbar. Es werden daher möglichst regelmäßige Packungen mit Teilchen von besonders kleinem Durchmesser verwendet.
Zur Ermittlung der Trennstufenhöhe in Abhängigkeit von der Fließgeschwindigkeit des Eluenten kann die sog. Van-Deemter-Gleichung für die Hochleistungsflüssigkeitschromatographie herangezogen werden:

wobei:
-
die Trennstufenhöhe,
 die lineare Fließgeschwindigkeit ist.
die lineare Fließgeschwindigkeit ist. -Term
berücksichtigt die Eddy-Diffusion,
die durch unterschiedliche Fließstrecken durch die Packung entsteht. Es gilt:
-Term
berücksichtigt die Eddy-Diffusion,
die durch unterschiedliche Fließstrecken durch die Packung entsteht. Es gilt:
 wobei
wobei
 den Packungsfaktor,
den Packungsfaktor, den Teilchendurchmesser bezeichnet.
den Teilchendurchmesser bezeichnet.
- Der
 -Term
berücksichtigt die longitudinale Diffusion. Die longitudinale Diffusion ist
die Diffusion der Analytenmoleküle in beide Richtungen der Trennstufe. Es
gilt:
-Term
berücksichtigt die longitudinale Diffusion. Die longitudinale Diffusion ist
die Diffusion der Analytenmoleküle in beide Richtungen der Trennstufe. Es
gilt:  wobei:
wobei:
 die Diffusionskonstante in der mobilen Phase und
die Diffusionskonstante in der mobilen Phase und-
der Labyrinthfaktor ist. Der Labyrinthfaktor berücksichtigt die
Porenstruktur der stationären Phase.
- der
 -Term
berücksichtigt die Peakverbreiterung
durch die langsame Gleichgewichtseinstellung zwischen der mobilen und der
stationären Phase. Hierbei ist noch die Diffusionskonstante
-Term
berücksichtigt die Peakverbreiterung
durch die langsame Gleichgewichtseinstellung zwischen der mobilen und der
stationären Phase. Hierbei ist noch die Diffusionskonstante  entlang der Poren der stationären Phase zu beachten. Es gilt
entlang der Poren der stationären Phase zu beachten. Es gilt 
Peaksymmetrie
Theoretisch sollte jede Substanz eine Chromatographiesäule als scharf eluierende Linie verlassen. Aus verschiedenen Gründen besitzen chromatographische Peaks jedoch immer eine gewisse Breite. Im Idealfall weisen sie dabei die Form einer Gauß’schen Glockenkurve auf. In der Praxis kommt es aber häufig vor, dass die Peaks von dieser Idealform abweichen und mehr oder weniger asymmetrisch erscheinen. Eine Asymmetrie, bei der der Frontanstieg des Peaks steiler ist als der Peakabfall, bezeichnet man als „Tailing“, während der Effekt, dass der Anstieg weniger steil ist als der Abfall als „Fronting“ oder auch „Leading“ bezeichnet wird. Der Tailingfaktor, der ein Maß für die Peaksymmetrie darstellt, wird bestimmt, indem man vom Peakmaximum das Lot zur Basislinie fällt, und in einer bestimmten Höhe, meist in 10 % der Peakhöhe, die Abstände zur Peakfront (a) und zum Peakende (b) ermittelt. Anschließend wird der Quotient der beiden Werte gebildet, wobei unterschiedliche Berechnungsformeln (z.B. nach IUPAC oder nach USP) in Gebrauch sind:
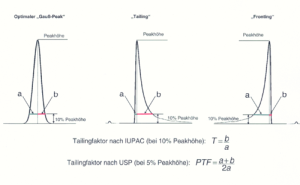
Ein idealer „Gauß-Peak“ erreicht dabei den Wert 1, Werte über 1 bedeuten „Tailing“, Werte unter 1 dagegen „Fronting“.
Verfahren
Literatur
- Karl Kaltenböck: Chromatographie für Einsteiger, Weinheim : Wiley-VCH-Verlag, Weinheim 2008, ISBN 978-3-527-32119-3.
- Andreas Heintz: Thermodynamik der Mischungen. Springer-Verlag GmbH, Deutschland 2017, ISBN 978-3-662-49923-8.


© biancahoegel.de
Datum der letzten Änderung: Jena, den: 23.06. 2025